Home / Alcohols – Acidity and Basicity
Alcohols, Epoxides and Ethers
Alcohols – Acidity and Basicity
Last updated: July 5th, 2025 |
Acid-Base Reactions Of Alcohols
Alcohols are mild acids. Typical aliphatic (i.e. “alkyl”) alcohols such as ethanol, isopropanol, and t-butanol have a pKa of about 16-18, making them slightly more acidic than water.
- Alcohols that are in conjugation with a pi bond or aromatic ring will be more acidic since the conjugate base is resonance-stabilized. One key example is phenol (C6H5OH). (pKa = 10).
- Nearby electron-withdrawing groups will stabilize the negative charge of the conjugate base through inductive effects. For example, 2,2,2-trifluoroethanol (pKa = 12) is considerably more acidic than ethanol (pKa = 16).
Alcohols are also weak bases. They can react with strong acids to give oxonium ions which have a pKa of about -2.
One of the keys to the reactions of alcohols as we go forward is that the conjugate acid is a better leaving group and the conjugate base is a better nucleophile.
Table of Contents
- Four Key Points To Review About Acid-Base Reactions
- Favorable and Unfavorable Acid-Base Reactions of Alcohols (2 Examples)
- Reviewing The Key Factors Which Determine Acidity
- Applying These Factors To The Acidity of Alcohols
- A Practice Question
- Summary: Acidity and Basicity of Alcohols
- Quiz Yourself!
1. Four Key Points To Review About Acid Base Reactions
- Every acid-base reaction has 4 components: an acid, a base, a conjugate acid, and a conjugate base.When an acid loses a proton, it becomes its conjugate base. When a base gains a proton, it becomes its conjugate acid. As mentioned in the previous post, the conjugate bas of an alcohol is called an alkoxide. The conjugate acid of an alcohol is called an oxonium ion. (See post: Acid-Base Reactions in Organic Chemistry)
- We usually describe acid-base reactions as an equilibrium. In acid-base reactions, the equilibrium will favor the direction where a stronger acid and stronger base produces a weaker acid and a weaker base.When you add HCl to NaOH, a violent acid-base reaction occurs, which leads to the formation of H2O (a weaker acid than HCl) and NaCl (a weaker base than NaOH). As you’ve no doubt discovered when adding table salt (NaCl) to water, this reaction doesn’t proceed to any significant extent in the reverse direction.
- We measure acidity using a term called pKa. This is a measure of the equilibrium constant for a species giving up a proton to form its conjugate base. pKa is on a scale of about -10 to 50. Sixty orders of magnitude! The higher the pKa the less acidic it is. Lower pKa (more negative ) = more acidic.
Water (pKa of 14.0) is a weaker acid than HCl (pKa of -8). (See article: How To Use a pKa Table) - The stronger the acid, the weaker the conjugate base. The weaker the acid, the stronger the conjugate base. The conjugate base of the strong acid HCl (pKa -8) is the innocuous chloride ion (Cl-), a very weak base. The conjugate base of the weak acid H2O (pKa 14) is the strongly basic hydroxide ion (HO-).
2. Examples of Acid-Base Reactions Of Alcohols
Here’s an example of a favorable acid-base reaction of alcohols. Note how we’re going from a stronger acid and stronger base to a weaker acid and weaker base [pKa values tell us for sure]
Here, deprotonation is very favourable. Note that the conjugate base of an alcohol is called an alkoxide.
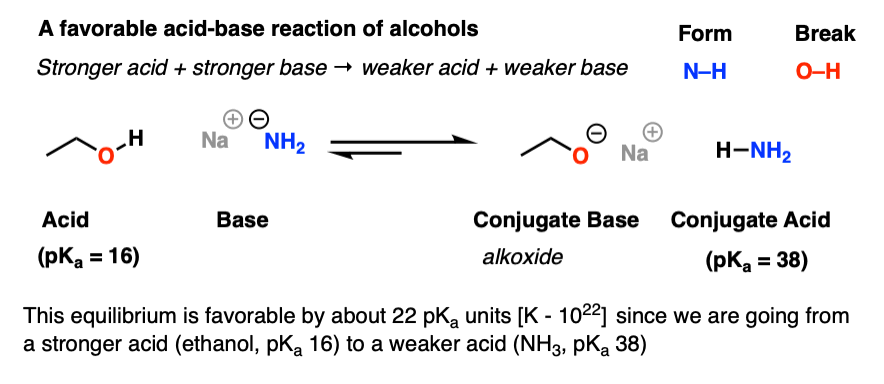
Here’s an example of a (very) unfavorable acid-base reaction of alcohols: protonation of an alcohol by NH3. The most important reason why this is unfavourable is because we’re going from a weaker acid (pKa 38) and weaker base to a stronger acid (pKa -2) and stronger base. The equilibrium constant is about 40 orders of magnitude in the wrong direction!
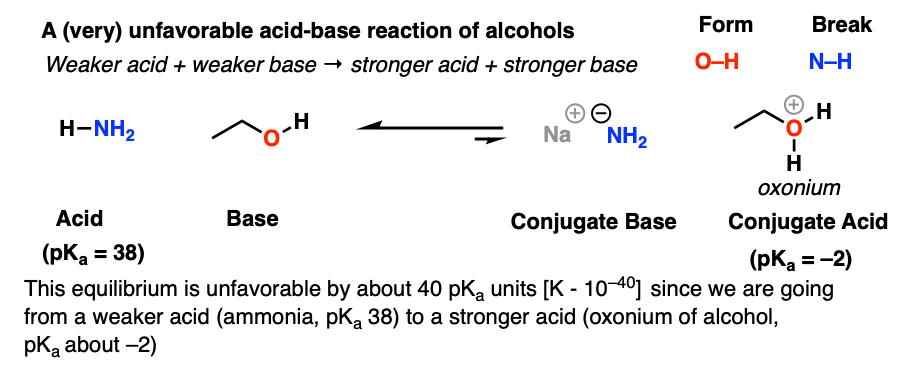
3. Reviewing The Key Factors That Determine Acidity
What determines how acidic a molecule is, anyway?
The key factor in determining acidity is the stability of the conjugate base. Any factor which makes the conjugate base more stable will increase the acidity of the acid. (See post: 5 Key Factors Which Influence Acidity)
What does that mean, exactly? Usually, it means stabilizing negative charge since the conjugate base will always be one unit of charge more “negative” than the acid. (See post: 7 Factors That Stabilize Negative Charge)
How is negative charge stabilized? Two ways.
- First, by bringing the charge closer to the positively charged nucleus [“opposite charges attract”, remember]. Across a row of the periodic table, for example, basicity decreases as we go from H3C– to H2N– to HO– to F– because the electronegativity of the atom is increasing. That negative charge is being held closer to the nucleus, and therefore is more stable. A good rule of thumb is, “the more stable a lone pair, the less basic it is. This is also why certain species are made acidic by adjacent electron-withdrawing groups.
- Second, by spreading charge out over a larger volume. Diffuse charge is more stable than concentrated charge. Down a row of the periodic table, for example, basicity decreases as we go from F– to Cl– to Br– to I– because that negative charge is being spread out over a larger volume (larger atoms). The larger atoms are said to be more “polarizable”. [Note that this effect dominates rather than electronegativity in this case.] This is also why resonance serves to stabilize charges; the charge is being spread across multiple atoms, therefore reducing individual charge density. (See post: In Summary – Resonance)
4. Applying These Principles To The Acidity Of Alcohols
How do these principles relate to alcohols? It’s quite simple, actually. Since we’ll always be comparing the same atom (oxygen) we don’t need to worry about periodic trends, and we just need to focus on resonance and adjacent electron-withdrawing groups.
Alcohols where the conjugate base is resonance stabilized will be more acidic. The classic example is cyclohexanol and phenol.
Cyclohexanol has the pKa of a typical alcohol (about 16). The pKa of phenol, however, is about 10. Let’s look:
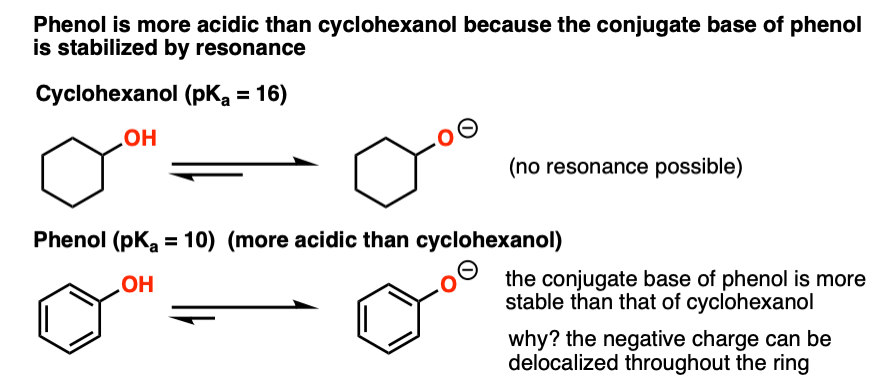
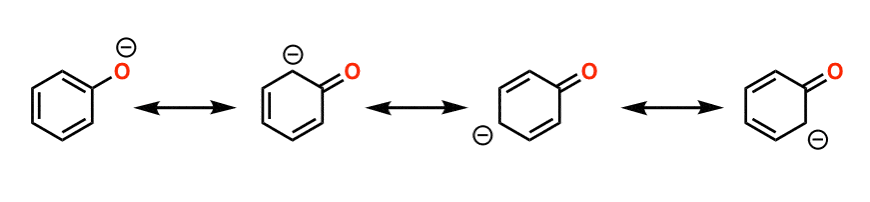
See how that negative charge on the oxygen of phenol can be “delocalized” back into the ring? That means the charge can be spread out throughout the molecule, which is stabilizing. Any factor which stabilizes the conjugate base will increase acidity.
Here’s another example. Compare ethanol (pKa 16) to 2,2,2-trifluoroethanol (pKa about 12). Why do you think trifluoroethanol is more acidic?
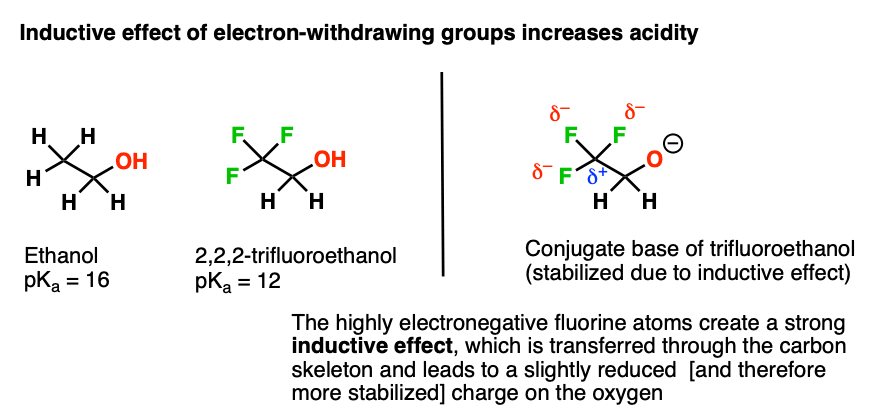
Compare their conjugate bases. What is fluorine doing here to make the conjugate base more stable?
This is an example of an inductive effect. Fluorine, being highly electronegative, pulls electron density away from the neighbouring carbon. That carbon, now being electron poor, pulls electron density away from the carbon next door. And that carbon, being slightly electron poor, can pull some electron density away from the oxygen.
The net result is that the oxygen has lower electron density, which is stabilizing. Again, stabilize the conjugate base –> increase acidity.
(See post: The Stronger The Acid, The Weaker The Conjugate Base)
This also works if we compare alcohol variations where we change the distance between the OH and the fluorine atom.
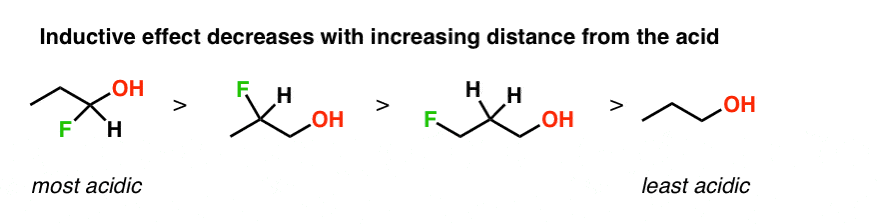
That’s because the inductive effect decreases in magnitude the farther away we go from the electronegative atom.
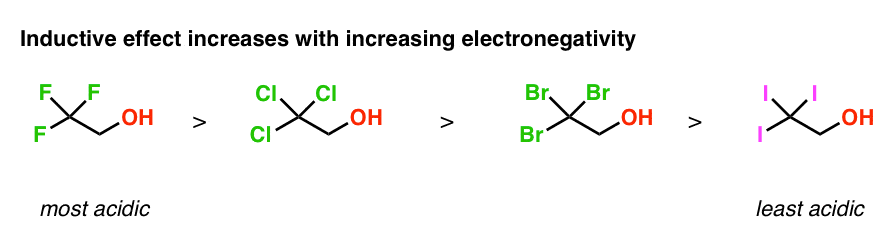
We can also use electronegativity trends to determine the order of acidity in these molecules. Since fluorine is more electronegative than chlorine which is more electronegative than bromine which is more electronegative than iodine, the inductive effect will be highest for CF3 and lowest for CI3.
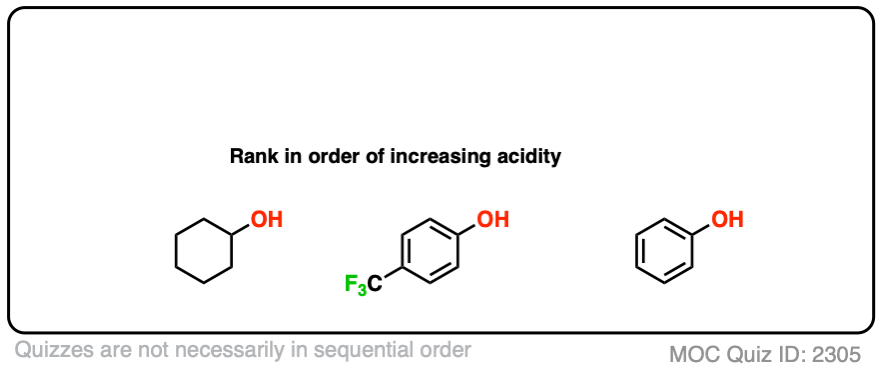
5. A Practice Question
Finally, one last example. We can even think of examples where these two effects are combined:
 Click to Flip
Click to Flip
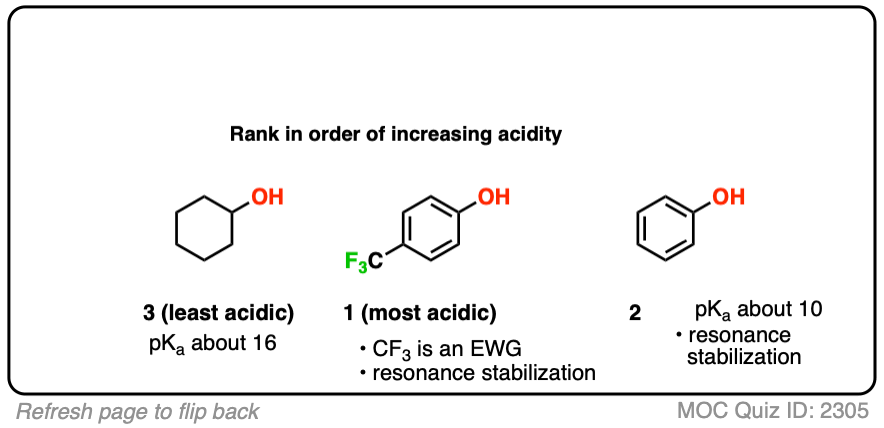
Which do you think might be most acidic here?
6. Summary: Acidity Of Alcohols
Now that we’ve covered the key factors governing the acidity of alcohols, we’re more prepared to get into the nitty gritty of their different reactions. In the next post we’ll start discussing how acidity and basicity affects the reaction conditions we can use.
For alcohols, since we’re always dealing with oxygen, the only relevant factors here are resonance and electron withdrawing groups.
Next Post – The Williamson Ether Synthesis
Notes
Related Articles
- The Williamson Ether Synthesis
- How to Use a pKa Table
- Acid-Base Reactions: Introducing Ka and pKa
- The Stronger The Acid, The Weaker The Conjugate Base
- Five Key Factors That Influence Acidity
- 7 Factors that stabilize negative charge in organic chemistry
- What makes a good leaving group?
- The Stronger The Acid, The Weaker The Conjugate Base
- Alcohols Can Act As Acids Or Bases (And Why It Matters)
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
1. Collected Acidity-Basicity Data
This website from the University of Estonia has a large curated list of studies on the acidity and basicity of various organic compounds.
Here is a leading reference. These pKa values refer to acetonitrile as solvent, so will be substantially different from those measured in aqueous solution, although the overall trends will be the same.
2. Strengths of Acids in Acetonitrile
A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg, I. Leito
Eur. J. Org. Chem. 2021, 2021, 1407.
DOI: doi.org/10.1002/ejoc.202001649
(Open access)
00 General Chemistry Review
01 Bonding, Structure, and Resonance
- How Do We Know Methane (CH4) Is Tetrahedral?
- Hybrid Orbitals and Hybridization
- How To Determine Hybridization: A Shortcut
- Orbital Hybridization And Bond Strengths
- Sigma bonds come in six varieties: Pi bonds come in one
- A Key Skill: How to Calculate Formal Charge
- The Four Intermolecular Forces and How They Affect Boiling Points
- 3 Trends That Affect Boiling Points
- How To Use Electronegativity To Determine Electron Density (and why NOT to trust formal charge)
- Introduction to Resonance
- How To Use Curved Arrows To Interchange Resonance Forms
- Evaluating Resonance Forms (1) - The Rule of Least Charges
- How To Find The Best Resonance Structure By Applying Electronegativity
- Evaluating Resonance Structures With Negative Charges
- Evaluating Resonance Structures With Positive Charge
- Exploring Resonance: Pi-Donation
- Exploring Resonance: Pi-acceptors
- In Summary: Evaluating Resonance Structures
- Drawing Resonance Structures: 3 Common Mistakes To Avoid
- How to apply electronegativity and resonance to understand reactivity
- Bond Hybridization Practice
- Structure and Bonding Practice Quizzes
- Resonance Structures Practice
02 Acid Base Reactions
- Introduction to Acid-Base Reactions
- Acid Base Reactions In Organic Chemistry
- The Stronger The Acid, The Weaker The Conjugate Base
- Walkthrough of Acid-Base Reactions (3) - Acidity Trends
- Five Key Factors That Influence Acidity
- Acid-Base Reactions: Introducing Ka and pKa
- How to Use a pKa Table
- The pKa Table Is Your Friend
- A Handy Rule of Thumb for Acid-Base Reactions
- Acid Base Reactions Are Fast
- pKa Values Span 60 Orders Of Magnitude
- How Protonation and Deprotonation Affect Reactivity
- Acid Base Practice Problems
03 Alkanes and Nomenclature
- Meet the (Most Important) Functional Groups
- Condensed Formulas: Deciphering What the Brackets Mean
- Hidden Hydrogens, Hidden Lone Pairs, Hidden Counterions
- Don't Be Futyl, Learn The Butyls
- Primary, Secondary, Tertiary, Quaternary In Organic Chemistry
- Branching, and Its Affect On Melting and Boiling Points
- The Many, Many Ways of Drawing Butane
- Wedge And Dash Convention For Tetrahedral Carbon
- Common Mistakes in Organic Chemistry: Pentavalent Carbon
- Table of Functional Group Priorities for Nomenclature
- Summary Sheet - Alkane Nomenclature
- Organic Chemistry IUPAC Nomenclature Demystified With A Simple Puzzle Piece Approach
- Boiling Point Quizzes
- Organic Chemistry Nomenclature Quizzes
04 Conformations and Cycloalkanes
- Staggered vs Eclipsed Conformations of Ethane
- Conformational Isomers of Propane
- Newman Projection of Butane (and Gauche Conformation)
- Introduction to Cycloalkanes
- Geometric Isomers In Small Rings: Cis And Trans Cycloalkanes
- Calculation of Ring Strain In Cycloalkanes
- Cycloalkanes - Ring Strain In Cyclopropane And Cyclobutane
- Cyclohexane Conformations
- Cyclohexane Chair Conformation: An Aerial Tour
- How To Draw The Cyclohexane Chair Conformation
- The Cyclohexane Chair Flip
- The Cyclohexane Chair Flip - Energy Diagram
- Substituted Cyclohexanes - Axial vs Equatorial
- Ranking The Bulkiness Of Substituents On Cyclohexanes: "A-Values"
- Cyclohexane Chair Conformation Stability: Which One Is Lower Energy?
- Fused Rings - Cis-Decalin and Trans-Decalin
- Naming Bicyclic Compounds - Fused, Bridged, and Spiro
- Bredt's Rule (And Summary of Cycloalkanes)
- Newman Projection Practice
- Cycloalkanes Practice Problems
05 A Primer On Organic Reactions
- The Most Important Question To Ask When Learning a New Reaction
- Curved Arrows (for reactions)
- Nucleophiles and Electrophiles
- The Three Classes of Nucleophiles
- Nucleophilicity vs. Basicity
- What Makes A Good Nucleophile?
- What Makes A Good Leaving Group?
- 3 Factors That Stabilize Carbocations
- Equilibrium and Energy Relationships
- 7 Factors that stabilize negative charge in organic chemistry
- 7 Factors That Stabilize Positive Charge in Organic Chemistry
- What's a Transition State?
- Hammond's Postulate
- Learning Organic Chemistry Reactions: A Checklist (PDF)
- Introduction to Oxidative Cleavage Reactions
06 Free Radical Reactions
- Free Radical Reactions
- 3 Factors That Stabilize Free Radicals
- Bond Strengths And Radical Stability
- Free Radical Initiation: Why Is "Light" Or "Heat" Required?
- Initiation, Propagation, Termination
- Monochlorination Products Of Propane, Pentane, And Other Alkanes
- Selectivity In Free Radical Reactions
- Selectivity in Free Radical Reactions: Bromination vs. Chlorination
- Halogenation At Tiffany's
- Allylic Bromination
- Bonus Topic: Allylic Rearrangements
- In Summary: Free Radicals
- Synthesis (2) - Reactions of Alkanes
- Free Radicals Practice Quizzes
07 Stereochemistry and Chirality
- Types of Isomers: Constitutional Isomers, Stereoisomers, Enantiomers, and Diastereomers
- How To Draw The Enantiomer Of A Chiral Molecule
- How To Draw A Bond Rotation
- Introduction to Assigning (R) and (S): The Cahn-Ingold-Prelog Rules
- Assigning Cahn-Ingold-Prelog (CIP) Priorities (2) - The Method of Dots
- Enantiomers vs Diastereomers vs The Same? Two Methods For Solving Problems
- Assigning R/S To Newman Projections (And Converting Newman To Line Diagrams)
- How To Determine R and S Configurations On A Fischer Projection
- The Meso Trap
- Optical Rotation, Optical Activity, and Specific Rotation
- Optical Purity and Enantiomeric Excess
- What's a Racemic Mixture?
- Chiral Allenes And Chiral Axes
- Stereochemistry Practice Problems and Quizzes
08 Substitution Reactions
- Nucleophilic Substitution Reactions - Introduction
- Two Types of Nucleophilic Substitution Reactions
- The SN2 Mechanism
- Why the SN2 Reaction Is Powerful
- The SN1 Mechanism
- The Conjugate Acid Is A Better Leaving Group
- Comparing the SN1 and SN2 Reactions
- Polar Protic? Polar Aprotic? Nonpolar? All About Solvents
- Steric Hindrance is Like a Fat Goalie
- Common Blind Spot: Intramolecular Reactions
- Substitution Practice - SN1
- Substitution Practice - SN2
09 Elimination Reactions
- Elimination Reactions (1): Introduction And The Key Pattern
- Elimination Reactions (2): The Zaitsev Rule
- Elimination Reactions Are Favored By Heat
- Two Elimination Reaction Patterns
- The E1 Reaction
- The E2 Mechanism
- E1 vs E2: Comparing the E1 and E2 Reactions
- Antiperiplanar Relationships: The E2 Reaction and Cyclohexane Rings
- Bulky Bases in Elimination Reactions
- Comparing the E1 vs SN1 Reactions
- Elimination (E1) Reactions With Rearrangements
- E1cB - Elimination (Unimolecular) Conjugate Base
- Elimination (E1) Practice Problems And Solutions
- Elimination (E2) Practice Problems and Solutions
10 Rearrangements
11 SN1/SN2/E1/E2 Decision
- Identifying Where Substitution and Elimination Reactions Happen
- Deciding SN1/SN2/E1/E2 (1) - The Substrate
- Deciding SN1/SN2/E1/E2 (2) - The Nucleophile/Base
- SN1 vs E1 and SN2 vs E2 : The Temperature
- Deciding SN1/SN2/E1/E2 - The Solvent
- Wrapup: The Key Factors For Determining SN1/SN2/E1/E2
- Alkyl Halide Reaction Map And Summary
- SN1 SN2 E1 E2 Practice Problems
12 Alkene Reactions
- E and Z Notation For Alkenes (+ Cis/Trans)
- Alkene Stability
- Alkene Addition Reactions: "Regioselectivity" and "Stereoselectivity" (Syn/Anti)
- Stereoselective and Stereospecific Reactions
- Hydrohalogenation of Alkenes and Markovnikov's Rule
- Hydration of Alkenes With Aqueous Acid
- Rearrangements in Alkene Addition Reactions
- Halogenation of Alkenes and Halohydrin Formation
- Oxymercuration Demercuration of Alkenes
- Hydroboration Oxidation of Alkenes
- m-CPBA (meta-chloroperoxybenzoic acid)
- OsO4 (Osmium Tetroxide) for Dihydroxylation of Alkenes
- Palladium on Carbon (Pd/C) for Catalytic Hydrogenation of Alkenes
- Cyclopropanation of Alkenes
- A Fourth Alkene Addition Pattern - Free Radical Addition
- Alkene Reactions: Ozonolysis
- Summary: Three Key Families Of Alkene Reaction Mechanisms
- Synthesis (4) - Alkene Reaction Map, Including Alkyl Halide Reactions
- Alkene Reactions Practice Problems
13 Alkyne Reactions
- Acetylides from Alkynes, And Substitution Reactions of Acetylides
- Partial Reduction of Alkynes With Lindlar's Catalyst
- Partial Reduction of Alkynes With Na/NH3 To Obtain Trans Alkenes
- Alkyne Hydroboration With "R2BH"
- Hydration and Oxymercuration of Alkynes
- Hydrohalogenation of Alkynes
- Alkyne Halogenation: Bromination and Chlorination of Alkynes
- Oxidation of Alkynes With O3 and KMnO4
- Alkenes To Alkynes Via Halogenation And Elimination Reactions
- Alkynes Are A Blank Canvas
- Synthesis (5) - Reactions of Alkynes
- Alkyne Reactions Practice Problems With Answers
14 Alcohols, Epoxides and Ethers
- Alcohols - Nomenclature and Properties
- Alcohols Can Act As Acids Or Bases (And Why It Matters)
- Alcohols - Acidity and Basicity
- The Williamson Ether Synthesis
- Ethers From Alkenes, Tertiary Alkyl Halides and Alkoxymercuration
- Alcohols To Ethers via Acid Catalysis
- Cleavage Of Ethers With Acid
- Epoxides - The Outlier Of The Ether Family
- Opening of Epoxides With Acid
- Epoxide Ring Opening With Base
- Making Alkyl Halides From Alcohols
- Tosylates And Mesylates
- PBr3 and SOCl2
- Elimination Reactions of Alcohols
- Elimination of Alcohols To Alkenes With POCl3
- Alcohol Oxidation: "Strong" and "Weak" Oxidants
- Demystifying The Mechanisms of Alcohol Oxidations
- Protecting Groups For Alcohols
- Thiols And Thioethers
- Calculating the oxidation state of a carbon
- Oxidation and Reduction in Organic Chemistry
- Oxidation Ladders
- SOCl2 Mechanism For Alcohols To Alkyl Halides: SN2 versus SNi
- Alcohol Reactions Roadmap (PDF)
- Alcohol Reaction Practice Problems
- Epoxide Reaction Quizzes
- Oxidation and Reduction Practice Quizzes
15 Organometallics
- What's An Organometallic?
- Formation of Grignard and Organolithium Reagents
- Organometallics Are Strong Bases
- Reactions of Grignard Reagents
- Protecting Groups In Grignard Reactions
- Synthesis Problems Involving Grignard Reagents
- Grignard Reactions And Synthesis (2)
- Organocuprates (Gilman Reagents): How They're Made
- Gilman Reagents (Organocuprates): What They're Used For
- The Heck, Suzuki, and Olefin Metathesis Reactions (And Why They Don't Belong In Most Introductory Organic Chemistry Courses)
- Reaction Map: Reactions of Organometallics
- Grignard Practice Problems
16 Spectroscopy
- Degrees of Unsaturation (or IHD, Index of Hydrogen Deficiency)
- Conjugation And Color (+ How Bleach Works)
- Introduction To UV-Vis Spectroscopy
- UV-Vis Spectroscopy: Absorbance of Carbonyls
- UV-Vis Spectroscopy: Practice Questions
- Bond Vibrations, Infrared Spectroscopy, and the "Ball and Spring" Model
- Infrared Spectroscopy: A Quick Primer On Interpreting Spectra
- IR Spectroscopy: 4 Practice Problems
- 1H NMR: How Many Signals?
- Homotopic, Enantiotopic, Diastereotopic
- Diastereotopic Protons in 1H NMR Spectroscopy: Examples
- 13-C NMR - How Many Signals
- Liquid Gold: Pheromones In Doe Urine
- Natural Product Isolation (1) - Extraction
- Natural Product Isolation (2) - Purification Techniques, An Overview
- Structure Determination Case Study: Deer Tarsal Gland Pheromone
17 Dienes and MO Theory
- What To Expect In Organic Chemistry 2
- Are these molecules conjugated?
- Conjugation And Resonance In Organic Chemistry
- Bonding And Antibonding Pi Orbitals
- Molecular Orbitals of The Allyl Cation, Allyl Radical, and Allyl Anion
- Pi Molecular Orbitals of Butadiene
- Reactions of Dienes: 1,2 and 1,4 Addition
- Thermodynamic and Kinetic Products
- More On 1,2 and 1,4 Additions To Dienes
- s-cis and s-trans
- The Diels-Alder Reaction
- Cyclic Dienes and Dienophiles in the Diels-Alder Reaction
- Stereochemistry of the Diels-Alder Reaction
- Exo vs Endo Products In The Diels Alder: How To Tell Them Apart
- HOMO and LUMO In the Diels Alder Reaction
- Why Are Endo vs Exo Products Favored in the Diels-Alder Reaction?
- Diels-Alder Reaction: Kinetic and Thermodynamic Control
- The Retro Diels-Alder Reaction
- The Intramolecular Diels Alder Reaction
- Regiochemistry In The Diels-Alder Reaction
- The Cope and Claisen Rearrangements
- Electrocyclic Reactions
- Electrocyclic Ring Opening And Closure (2) - Six (or Eight) Pi Electrons
- Diels Alder Practice Problems
- Molecular Orbital Theory Practice
18 Aromaticity
- Introduction To Aromaticity
- Rules For Aromaticity
- Huckel's Rule: What Does 4n+2 Mean?
- Aromatic, Non-Aromatic, or Antiaromatic? Some Practice Problems
- Antiaromatic Compounds and Antiaromaticity
- The Pi Molecular Orbitals of Benzene
- The Pi Molecular Orbitals of Cyclobutadiene
- Frost Circles
- Aromaticity Practice Quizzes
19 Reactions of Aromatic Molecules
- Electrophilic Aromatic Substitution: Introduction
- Activating and Deactivating Groups In Electrophilic Aromatic Substitution
- Electrophilic Aromatic Substitution - The Mechanism
- Ortho-, Para- and Meta- Directors in Electrophilic Aromatic Substitution
- Understanding Ortho, Para, and Meta Directors
- Why are halogens ortho- para- directors?
- Disubstituted Benzenes: The Strongest Electron-Donor "Wins"
- Electrophilic Aromatic Substitutions (1) - Halogenation of Benzene
- Electrophilic Aromatic Substitutions (2) - Nitration and Sulfonation
- EAS Reactions (3) - Friedel-Crafts Acylation and Friedel-Crafts Alkylation
- Intramolecular Friedel-Crafts Reactions
- Nucleophilic Aromatic Substitution (NAS)
- Nucleophilic Aromatic Substitution (2) - The Benzyne Mechanism
- Reactions on the "Benzylic" Carbon: Bromination And Oxidation
- The Wolff-Kishner, Clemmensen, And Other Carbonyl Reductions
- More Reactions on the Aromatic Sidechain: Reduction of Nitro Groups and the Baeyer Villiger
- Aromatic Synthesis (1) - "Order Of Operations"
- Synthesis of Benzene Derivatives (2) - Polarity Reversal
- Aromatic Synthesis (3) - Sulfonyl Blocking Groups
- Birch Reduction
- Synthesis (7): Reaction Map of Benzene and Related Aromatic Compounds
- Aromatic Reactions and Synthesis Practice
- Electrophilic Aromatic Substitution Practice Problems
20 Aldehydes and Ketones
- What's The Alpha Carbon In Carbonyl Compounds?
- Nucleophilic Addition To Carbonyls
- Aldehydes and Ketones: 14 Reactions With The Same Mechanism
- Sodium Borohydride (NaBH4) Reduction of Aldehydes and Ketones
- Grignard Reagents For Addition To Aldehydes and Ketones
- Wittig Reaction
- Hydrates, Hemiacetals, and Acetals
- Imines - Properties, Formation, Reactions, and Mechanisms
- All About Enamines
- Breaking Down Carbonyl Reaction Mechanisms: Reactions of Anionic Nucleophiles (Part 2)
- Aldehydes Ketones Reaction Practice
21 Carboxylic Acid Derivatives
- Nucleophilic Acyl Substitution (With Negatively Charged Nucleophiles)
- Addition-Elimination Mechanisms With Neutral Nucleophiles (Including Acid Catalysis)
- Basic Hydrolysis of Esters - Saponification
- Transesterification
- Proton Transfer
- Fischer Esterification - Carboxylic Acid to Ester Under Acidic Conditions
- Lithium Aluminum Hydride (LiAlH4) For Reduction of Carboxylic Acid Derivatives
- LiAlH[Ot-Bu]3 For The Reduction of Acid Halides To Aldehydes
- Di-isobutyl Aluminum Hydride (DIBAL) For The Partial Reduction of Esters and Nitriles
- Amide Hydrolysis
- Thionyl Chloride (SOCl2) And Conversion of Carboxylic Acids to Acid Halides
- Diazomethane (CH2N2)
- Carbonyl Chemistry: Learn Six Mechanisms For the Price Of One
- Making Music With Mechanisms (PADPED)
- Carboxylic Acid Derivatives Practice Questions
22 Enols and Enolates
- Keto-Enol Tautomerism
- Enolates - Formation, Stability, and Simple Reactions
- Kinetic Versus Thermodynamic Enolates
- Aldol Addition and Condensation Reactions
- Reactions of Enols - Acid-Catalyzed Aldol, Halogenation, and Mannich Reactions
- Claisen Condensation and Dieckmann Condensation
- Decarboxylation
- The Malonic Ester and Acetoacetic Ester Synthesis
- The Michael Addition Reaction and Conjugate Addition
- The Robinson Annulation
- Haloform Reaction
- The Hell–Volhard–Zelinsky Reaction
- Enols and Enolates Practice Quizzes
23 Amines
- The Amide Functional Group: Properties, Synthesis, and Nomenclature
- Basicity of Amines And pKaH
- 5 Key Basicity Trends of Amines
- The Mesomeric Effect And Aromatic Amines
- Nucleophilicity of Amines
- Alkylation of Amines (Sucks!)
- Reductive Amination
- The Gabriel Synthesis
- Some Reactions of Azides
- The Hofmann Elimination
- The Hofmann and Curtius Rearrangements
- The Cope Elimination
- Protecting Groups for Amines - Carbamates
- The Strecker Synthesis of Amino Acids
- Introduction to Peptide Synthesis
- Reactions of Diazonium Salts: Sandmeyer and Related Reactions
- Amine Practice Questions
24 Carbohydrates
- D and L Notation For Sugars
- Pyranoses and Furanoses: Ring-Chain Tautomerism In Sugars
- What is Mutarotation?
- Reducing Sugars
- The Big Damn Post Of Carbohydrate-Related Chemistry Definitions
- The Haworth Projection
- Converting a Fischer Projection To A Haworth (And Vice Versa)
- Reactions of Sugars: Glycosylation and Protection
- The Ruff Degradation and Kiliani-Fischer Synthesis
- Isoelectric Points of Amino Acids (and How To Calculate Them)
- Carbohydrates Practice
- Amino Acid Quizzes
25 Fun and Miscellaneous
- A Gallery of Some Interesting Molecules From Nature
- Screw Organic Chemistry, I'm Just Going To Write About Cats
- On Cats, Part 1: Conformations and Configurations
- On Cats, Part 2: Cat Line Diagrams
- On Cats, Part 4: Enantiocats
- On Cats, Part 6: Stereocenters
- Organic Chemistry Is Shit
- The Organic Chemistry Behind "The Pill"
- Maybe they should call them, "Formal Wins" ?
- Why Do Organic Chemists Use Kilocalories?
- The Principle of Least Effort
- Organic Chemistry GIFS - Resonance Forms
- Reproducibility In Organic Chemistry
- What Holds The Nucleus Together?
- How Reactions Are Like Music
- Organic Chemistry and the New MCAT
26 Organic Chemistry Tips and Tricks
- Common Mistakes: Formal Charges Can Mislead
- Partial Charges Give Clues About Electron Flow
- Draw The Ugly Version First
- Organic Chemistry Study Tips: Learn the Trends
- The 8 Types of Arrows In Organic Chemistry, Explained
- Top 10 Skills To Master Before An Organic Chemistry 2 Final
- Common Mistakes with Carbonyls: Carboxylic Acids... Are Acids!
- Planning Organic Synthesis With "Reaction Maps"
- Alkene Addition Pattern #1: The "Carbocation Pathway"
- Alkene Addition Pattern #2: The "Three-Membered Ring" Pathway
- Alkene Addition Pattern #3: The "Concerted" Pathway
- Number Your Carbons!
- The 4 Major Classes of Reactions in Org 1
- How (and why) electrons flow
- Grossman's Rule
- Three Exam Tips
- A 3-Step Method For Thinking Through Synthesis Problems
- Putting It Together
- Putting Diels-Alder Products in Perspective
- The Ups and Downs of Cyclohexanes
- The Most Annoying Exceptions in Org 1 (Part 1)
- The Most Annoying Exceptions in Org 1 (Part 2)
- The Marriage May Be Bad, But the Divorce Still Costs Money
- 9 Nomenclature Conventions To Know
- Nucleophile attacks Electrophile
27 Case Studies of Successful O-Chem Students
- Success Stories: How Corina Got The The "Hard" Professor - And Got An A+ Anyway
- How Helena Aced Organic Chemistry
- From a "Drop" To B+ in Org 2 – How A Hard Working Student Turned It Around
- How Serge Aced Organic Chemistry
- Success Stories: How Zach Aced Organic Chemistry 1
- Success Stories: How Kari Went From C– to B+
- How Esther Bounced Back From a "C" To Get A's In Organic Chemistry 1 And 2
- How Tyrell Got The Highest Grade In Her Organic Chemistry Course
- This Is Why Students Use Flashcards
- Success Stories: How Stu Aced Organic Chemistry
- How John Pulled Up His Organic Chemistry Exam Grades
- Success Stories: How Nathan Aced Organic Chemistry (Without It Taking Over His Life)
- How Chris Aced Org 1 and Org 2
- Interview: How Jay Got an A+ In Organic Chemistry
- How to Do Well in Organic Chemistry: One Student's Advice
- "America's Top TA" Shares His Secrets For Teaching O-Chem
- "Organic Chemistry Is Like..." - A Few Metaphors
- How To Do Well In Organic Chemistry: Advice From A Tutor
- Guest post: "I went from being afraid of tests to actually looking forward to them".
While some may think the difference in acidity of trichloro v trifluoroethanol is trivial, I think it is the norm. Can anyone give me one example in which a fluoro substituent is a better electron withdrawer than a chlorine, except for haloacetic acids. If you wish to refer to the haloacetic acids, then please explain the acidity of the substituents of substituted acetic acids as a class.
Correct me if I have the wrong person, but I believe you have been commenting on this very topic for over 10 years. Do you have an article that you might recommend I could direct my readers to so that they could be more fully informed on this point. Thank you.
This is the great conceptual website which give proper guidness and understanding about the reactions.greatly thanks sir .excellent work🤗🤩😍😍😍
Esterification-hydrolysis is definitely an equilibrium. If you do not drive the reaction you end up in the middle.
I agree that it is not a purely acid base reaction, but there is definitely an acid base component in it, and this is why you need an acid or a base to at least start it up.
But I see your point, If you have a simple acid base reaction in a close container it is correct.
I have a more serious question ( I am not a chemist ).
In the Fischer esterification reaction, the first step is a protonation at the oxygen of the carbonyl group. I think this step is highly unfavorable. It amount to move the equilibrium to an enol, or to consider the pKa of RCO2H2+ which would be a very very strong acid.
My first question is:
Why don’t we do the nucleophile attack FIRST, and the protonation AFTER on the side of the O-. It looks more favorable.
My second question is:
Chemists seems to have no problem making an alkoxy anion leave the tetrahedral intermediate, but it seems to be a big mistake to make an hydroxy anion leave. Why?
The pKa of their conjugate acid is about the same.
Note: Your web site is wonderful.
I am not convinced that we should learn:
Stronger Acid + Stronger Base –> Weaker Acid + Weaker Base
It depends on other factors.
For instance:
Hydrolysis of an ester. You start with water (pKa = 16) and an ester and end up with a carboxylic acid (pKa = 4) and an alcohol.
Or you leave a nice glass of wine on Friday night to find vinegar on Saturday morning.
Ester hydrolysis (and its opposite, Fischer esterification) are not acid-base equilibria. This post is about simple acid-base reactions where the only bonds forming and breaking are to hydrogen.
What is more acidic alkene or alcohol?
What do you think, based on a pKa table?
You say that, in acidity of alcohols; more stabilization of the alkoxide ion formed (by inductive effects, etc), means more acidic is the alcohol.
Sure, but the trend you would get is not analogous to the data found in the gas phase oppose the data found from the solutions.
The acidity of alcohols is mainly due to polarizibility and solvation.
Sources: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Alcohols/Properties_of_Alcohols/Acidities_of_Alcohols
AND
Organic Chemistry (Sixth Edition) by Robert Thornton Morrison & Robert Neilson Boyd [Pg. 228]
Hi – I make the assumption that we are dealing with solution-phase chemistry, not gas-phase. Any quoted pKa values are quoted in water and/or DMSO as solvent.
Question.. besides solvent influence how exactly does electronegativity suddenly take precedence over polarizability in stabilizing negative charge and increasing acidity of the hydroxyl function ?? I am confused here. I understand polarizability as taking precedence due stability of the distributed charge of the larger atom and can further justify polarizability and it’s effect of charge stabilization here through the fact of the longer time required for electrons to travel around the larger atom creating a larger dipole moment and therefore a greater partial charge. Yet starting a new sentence, the higher electronegativity (due higher attraction generated through closness of opposing charges) of the smaller halides mainly in question here can b justified to be electron withdrawing to carbon and oxygen there fore stabilizing the anion from both points being electronegativity, and polarizability.. which i guess answers my question but raises then where does polarizability actually take precedence ??
Any meaningful way to spread out the charge density (ex. resonance or “polarizability”) takes precedence.
For example, phenol (the right-most molecule in question before the conclusion above), has a pka of 10 due to resonance structures. That’s several hundred times more acidic than 2,2,2-trifluoroethanol, the alcohol with 3 fluorines used as an example for the inductive effect above. CF3 is a lot of inductive power, but resonance is still more important.
Another example for polarizability: primary alcohols (OH group) have a pka around 16 while primary thiols (SH group) have a pka of around 10 — a million times more acidic.
Just be careful if your “polarizability of an electron cloud” is referring to stabilization by e.g. alkyl chains (I’m pretty sure this is actually just hyperconjugation), as opposed to the “polarizability” stabilizing an iodide or sulfide conjugate base (pretty sure this is just a shell or size effect). The former effect is weak (+-1 pka) and its trend reverses in water vs gas phase (e.g. H2O > methanol > ethanol), because solvent effects overpower it. Trends from the latter effect (e.g. halogenic acid strength or H2O vs H2S) are not reversed between water and gas phase.
the order on your second to last rank-the-acidity is incorrect. it would be correct if electronegativity were the only player in ranking acidity, but it is not. the size of the halogen also factors in here…spreading out the charge increases acidity…i.e. why HF is a weak acid…<HCl< HBr < HI …consequently, your ranking of 2,2,2trifolorethanol should be reversed…2,2,2trifolorethanol (weakest) < 2,2,2tricholor etc… pka for triflorethanol is ~12.46..pka for tricholorethanol is ~12.02
OK, so I am curious here.. I posted a question regarding electronegativity vs polarizability. I can understand polarizability of the larger halogens generating the stronger acid through the creation of the larger dipole moment or the fact of the small halogen and it’s higher electronegativity (closeness of opposite charges) withdrawing electrons more strongly, but what conditions (possibly such as solvent) will determine reactivity of the species in concern ?? I see it said that polarizability takes presidence yet as you have stated he shows opposing acidity trends in the example as you have shown. Are there environmental conditions I am overlooking that aren’t stated to justify this ?? Or maybe the characteristics oh the double bonded oxygen contributing ??
The person you replied to is correct on one point and wrong on everything else. 2,2,2-trichloroethanol is indeed more acidic than 2,2,2-trifluoroethanol.
However, the difference is very small: 12.24 vs 12.46 pka, respectively.
Furthermore, aside from 2,2,2-trichloroethanol, the pattern is in fact correct. It should not be wholly reversed like the person you replied to stated. 2,2,2-tribromoethanol clocks in at 12.7 pka, in accordance with the pattern, and 2,2,2-triiodoethanol hasn’t been measured as far as I could find.
Finally, the mechanism at play here has nothing to do with the polarizability in the context you’re used to or anything related to the halogenic acids, HF, HCl, HBr, and HI. The polarizability of larger molecules is relevant for you in explaining intermolecular forces (induced dipole attractions). The stability of halogen anions (the conjugate base of a halogenic acid) has to do with the charge density. Fluorine is very small, so carrying a negative charge by itself concentrates the negative charge in a very small space alongside other electrons, which leads to a repulsive and destabilizing interaction. In iodine, the single extra electron is spread out over a much larger volume which minimizes destabilizing interactions. This along with orbital overlap (HSAB theory – traditionally covered in your first inorganic chemistry course) should more or less account for the differences in halogenic acid pka.
Honestly, just follow this page’s advice and ignore the technicality on 2,2,2-trichloroethanol. The difference of 0.3 pka is so trivially small that it would involve a complex analysis of a multitude of variables to pinpoint why, so there’s no way it pops up on an exam with the expectation you know about the exception of 2,2,2-trichloroethanol.
Thank you, Blaise!
what is the effect of the positive effect type of inductive effects on acidity of alcohol and basicity amine
Amazing. Reopen my mind of organic chem.
I have a question. Do you think the acidity/basicity is correlated to its 1H NMR chemical shift? Specifically for protonated amino acids.
Thanks.
Regarding the phenol vs cyclohexanol example, since most resonance forms break the aromaticity of the ring, is the charge delocalization still significant enough as to make it a more stable conjugate base?
I’m sorry but I have a question why primary >secondary >tertiary > alcohols in acidity order
Thank you
Think about how alkyl groups affect the electron-richness of the alcohol, relative to hydrogen. Are they electron donors or electron acceptors? Do they make it more electron rich or less electron rich? One of the key factors affecting acidity is the stability of the conjugate base, which in this case will be an alkoxide (O-). The oxygen already has a negative charge. What will electron donors do to that negative charge? Make it bigger or smaller? And what do you think is more stable, a larger charge, or a smaller charge? If the charge is less stable, that means that the conjugate base will be more basic, which means that the parent alcohol will be less acidic.
This is just an excellent write up. My organic chemistry textbook wasn’t even as detailed as this is.
Glad you found it useful Clarence!
thanku so much! it is really helping me get through my MCAT :)
Just one word for this-awesome! really helpful! u saved me man :-)
Thank you for sharing the article. It’s very interesting. I’d love to hear more from you.
I apologize if I’m wrong, but isn’t the pKa of ethanol closer to 16 (15.9 is the most cited value)?
Also, “the electronegativity of the nucleus is increasing” and “the negatively charged oxygen on trifluoroethanol will have some of its charge redistributed to fluorine” might be somewhat difficult to understand to undergraduate students. Maybe a little bit of elaboration and/or rephrasing might be in order.
Thank you once more. Fixed. I am in your debt for your excellent suggestions.
thank you mamid…..I am an undergraduate and honestly i could not understand some of the phrases being used to explain
what is effect of the positive effects on acidity of alcohol(OH)