Stereochemistry and Chirality
Optical Rotation, Optical Activity, and Specific Rotation
Last updated: June 2nd, 2026 |
Optical Rotation, Optical Activity, and Specific Rotation
If you’ve been learning about stereochemistry, enantiomers, and diastereomers, the following might sound familiar:
- Diastereomers have different physical properties (i.e. boiling points, melting points, solubilities)
- Enantiomers have identical physical properties* , with one exception: enantiomers rotate plane-polarized light in equal and opposite directions, which is why they are sometimes called “optical isomers”.
*(assuming an achiral environment)
[If you’re unclear on the difference between enantiomers and diastereomers, I’d suggest going back to this post]
What does that term “optical rotation” mean? Or, for that matter, “optical activity”? You might also have heard of “specific rotation”. What’s that? We’ll cover all of these concepts below.
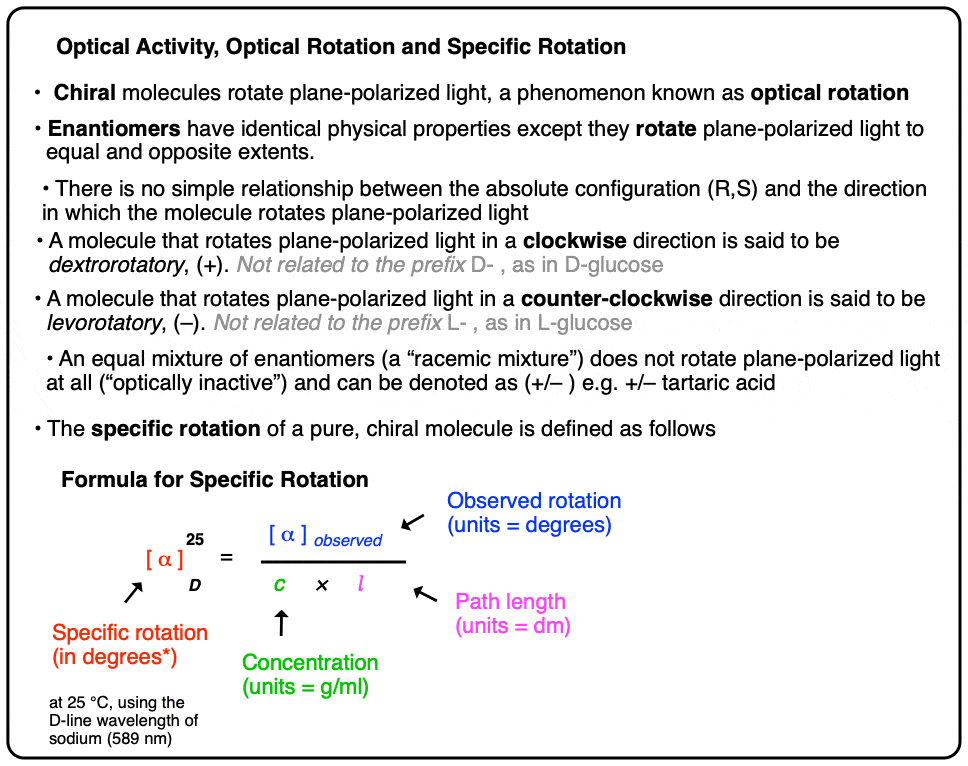
Table of Contents
- Louis Pasteur’s Discovery of “Left-Handed” and “Right-Handed” Tartaric Acid Crystals
- The Three Stereoisomers Of Tartaric Acid, And How They Are Related
- Do Molecules With An (R) Configuration always Rotate Plane-Polarized Light To The Right? (Hint: No)
- Untangling The Differences Between R and S, D- and L- , d- and l-, and (+)- and (–)-
- Polarimetry: The Measurement of Optical Rotation
- Specific Rotation: A Common Standard For Comparing Optical Rotation
- Specific Rotation: A Sample Problem
- Conclusion: Optical Rotation And Specific Rotation
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
This post was co-authored with Matt Pierce of Organic Chemistry Solutions. Ask Matt about scheduling an online tutoring session.
1. Louis Pasteur’s Discovery of “Left-Handed” and “Right-Handed” Tartaric Acid Crystals
Louis Pasteur is more than just the man whose name was lent to the process of “pasteurization”. He is also the father of organic stereochemistry . In 1848, Pasteur published a study on the recrystallization of various salts of tartaric acid, or “tartrates”, which are found naturally in wine (aka “wine diamonds”).

Of particular interest to Pasteur were the crystalline forms “racemic acid” (from the Latin racemus for “a bunch of grapes”), which at that time was thought to be an isomer of tartaric acid.
At the time, it was known that racemic acid did not turn the plane of polarized light, whereas “tartar”, the most common salt of tartaric acid, rotated plane-polarized light to the right [“dextrorotatory”, or (+) ]
Upon close inspection, Pasteur noticed that the potassium sodium salt of “racemic acid” crystallized in two separate crystal forms which were mirror images of each other. According to rules of crystal morphology, one type was “right handed”, and the other was “left handed” . [Maybe you’ve heard of “left-handed” and “right handed” screws? The process of naming left and right-handed crystals is similar. ]

This was a strange result, since there was no reason to think that crystals that did not themselves rotate plane-polarized light should have any chirality. Pasteur carefully arranged the crystals and discovered that one half of them were right handed and the other half were left handed. Taken into aqueous solution, the right-handed crystals were dextrorotatory (exactly like crystals of “tartar”, from wine) and the left-handed crystals were levorotatory, to precisely the same degree.
From this Pasteur postulated that the two molecules were mirror images of each other – even though it would be years before the absolute structure of tartaric acid was known, and 25 years before Van’t Hoff proposed the tetrahedral shape of carbon as a means of explaining the existence of optical isomers. [Note 2]
2. The Structure of The Three Stereoisomers Of Tartaric Acid
We now know that what Pasteur called “racemic acid” was not a single compound, but in fact a mixture of two enantiomers of tartrate. Upon crystallization, the [S,S] and [R,R] enantiomers gave different crystals which Pasteur separated mechanically, i.e. by hand. [Note: in the figure below, we show “tartaric acids”; Pasteur did his work on the salts of the conjugate bases, which we call “tartrates”]
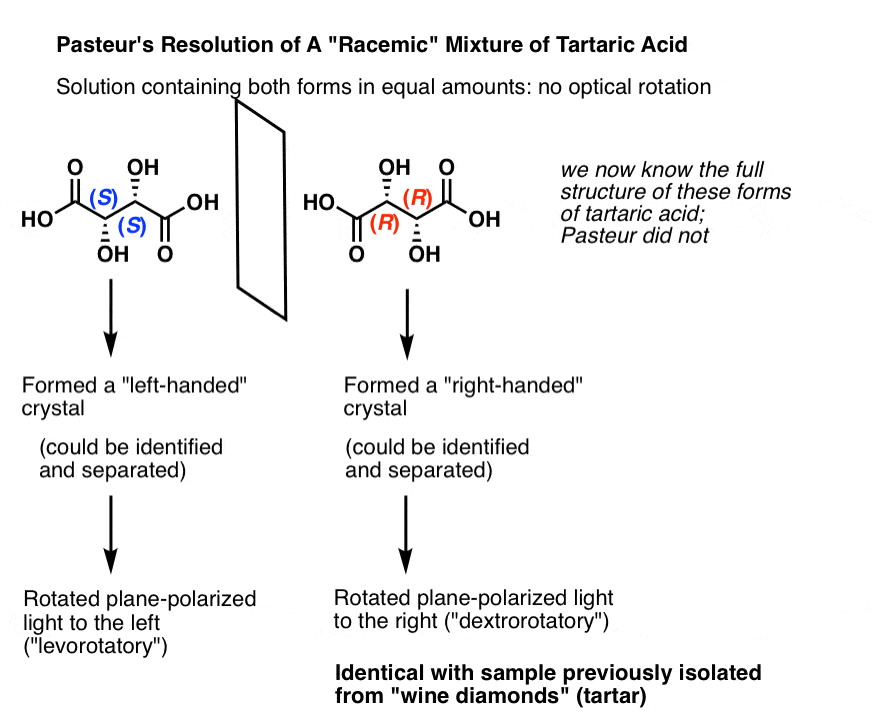
In isolation (S,S) tartaric acid rotates plane-polarized light to the left, and (R,R) tartaric acid rotates plane-polarized light to the right. Naturally occurring wine diamonds are (R,R) and thus dextrorotatory.
Thus these are called “optical isomers” in that they differ solely in the direction of their optical rotation.
By the way, Pasteur also studied a third form of tartaric acid that does not rotate plane-polarized light at all. This form was called “meso” (Greek for middle, since the light was rotated neither to the left nor the right). The configuration of the two chiral centers were subsequently determined to be (R,S).
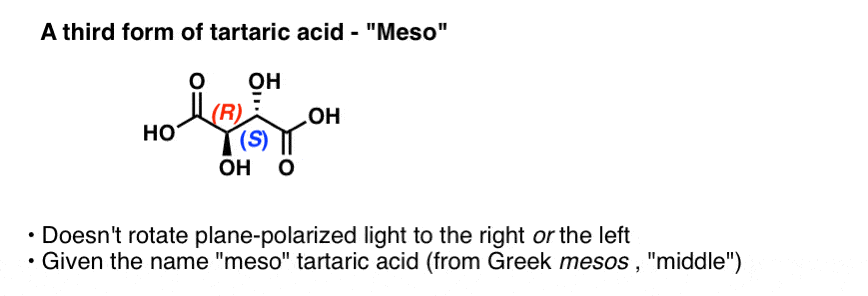
If you’ve covered chirality at all, this term “meso” might be familiar to you. Despite having two chiral enters, “meso” tartaric acid has an internal plane of symmetry and is therefore not a chiral molecule. The name “meso” has come to denote a whole class of compounds that bear chiral centers but are not themselves chiral.

3. Do Molecules With An (R) Configuration always Rotate Plane-Polarized Light To The Right? (Hint: No)
Sometimes you might see a molecule that rotates plane-polarized light to the right (dextrorotatory) described as (+) and a molecule that rotates plane-polarized light to the left (levorotatory) as (–).
Hence, we can have (+)-tartaric acid and (–)-tartaric acid, (+)-glucose and (–)-glucose, and (+)-morphine and (–)-morphine – all pairs of enantiomers.
We also noted that (+)-tartaric acid is (R,R) and (–) tartaric acid is (S,S).
All this begs a question. What is the relationship between the direction of optical rotation and the structure of a molecule? Are molecules with an (R) configuration always dextrorotatory, and molecules with an (S) configuration always levorotatory?
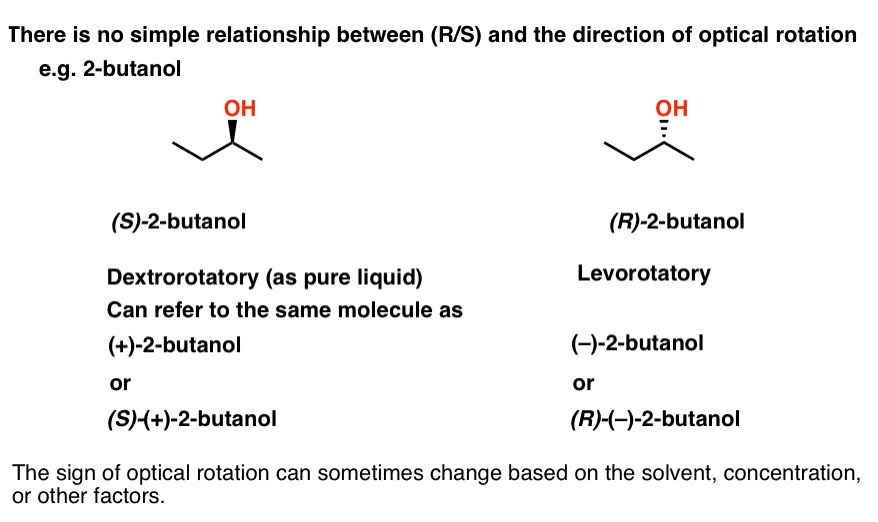
No! There is no simple way to predict the direction of rotation based on the structure. If you want to know what direction a molecule rotates polarized light, you just have to measure it.
For example, (S)-2-butanol is dextrorotatory (+)as a pure liquid, while (R)-2-butanol is levorotatory (–). If we wish, we could also describe (S)-2-butanol as (+)-2-butanol, or even (S)-(+)-2-butanol if you prefer.
4. Untangling The Differences Between R and S, D- and L- , d- and l-, and (+)- and (–)-
This of the problems with discussing a relatively old field like organic stereochemistry is that there are many layers of terminology, some obsolete, that must be peeled away.
The Cahn-Ingold-Prelog system [the origin of naming chiral centers (R) and (S) ] is a relatively new development, dating back to 1951.
(R) and (S) describe the absolute stereochemistry of chiral centers, which you can use to draw the molecule if you know the connectivity of a molecule and understand how to apply the system.
The (R,S) system only became possible once the absolute configuration of molecules could be confirmed, which itself only became possible with the development of X-ray crystallography. [Specifically, Bijouvet in 1951 determined the absolute structure of sodium rubidibum (+)-tartrate using the “heavy atom” method.]
Before the (R,S) system, we had the D, L- system, which were based on Emil Fischer’s guess of the absolute structure of (+)-glyceraldehyde, and then applied to other molecules through chemical analogy. [Note 1].
For example, the levorotatory (–) form of tartaric acid (S, S) is also sometimes described as D-tartaric acid for reasons we won’t go in to here, and conversely, the dextrorotary form (R, R) is described as L-tartaric acid. You see the terms D– and L– also used for amino acids; the essential amino acids are all L.
To add to the confusion, sometimes lowercase “d” and “l” are used to abbreviate “dextrorotatory” and “levorotatory” respectively in place of (+) and (–). So we have d-tartaric acid, which is (+), and l-tartaric acid, which is (–). If we have a mixture of the two (a racemic mixture) you might see this referred to as dl-tartaric acid. Note that IUPAC has designated these terms as obsolete – use (+)/(–) instead.

5. Polarimetry: The Measurement of Optical Rotation
Let’s briefly delve into what got us into this situation in the first place: the measurement of optical rotation. It has been known since at least the 1810s that certain crystals (e.g. quartz) had chiral forms that rotated plane-polarized light in equal and opposite directions. Furthermore, solutions of glucose and turpentine were measured using this technique and shown to be optically active.
Although the equipment has changed, the technique of polarimetry is no different than it was in Pasteur and Biot’s day. The first step is to pass light through a polarizer, which only allows light with waves aligned in one direction to pass. This polarized light is then transmitted through the material to be studied, in our case a cell containing a solution of the molecule.

At the other end, a second polarizer is rotated a given angle θ until this light is transmitted through the slit. Obviously if the solution is not optically active at all, this angle will be zero.
Here is a diagram of a modern polarimeter. Image source: wikipedia
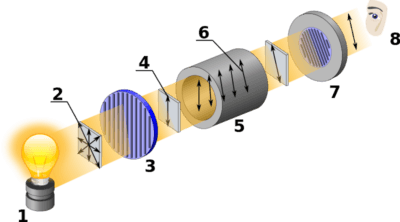
The technical details of how early scientists obtained polarized light are pretty fascinating. More detail here.
6. Specific Rotation: A Common Standard For Comparing Optical Rotation
Now comes the final piece of the puzzle: standardization. It would be useful to have a common standard for optical rotation that allowed us to compare samples collected under slightly different concentrations and path lengths, a little bit like how earned run average (ERA) allows for comparison of performance between pitchers, or goals against average (GAA) for goalies, or batting average, or quarterback passer rating… you can pick your own sports metaphor.
The term that has been developed for this is specific rotation.
The specific rotation of a molecule is the rotation in degrees observed upon passing polarized light through a path length of 1 decimetre (dm) at a concentration of 1 g/mL.
To convert an observed rotation to specific rotation, divide the observed rotation by the concentration in g/mL and the path length in decimeters (dm).
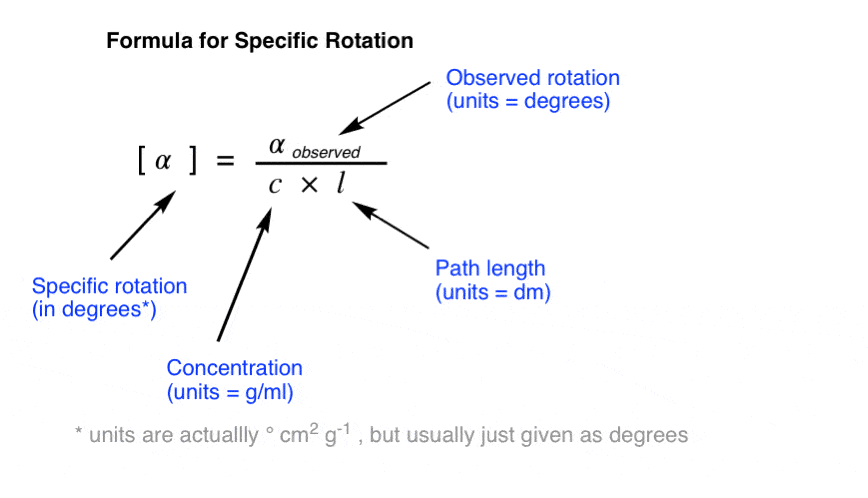
{kind=link}
NOTE: As commenter Dr. Fred points out, in most cases dissolving a whole gram of material into a millilitre of solvent is impractical! In laboratory settings, concentration is measured in g / 100 mL and a correction factor of 100 is applied to the numerator. Values of c from the literature should be assumed to be in g / 100 mL.
For reporting purposes, the specific rotation is usually accompanied by the wavelength (often the D-line of sodium, 589 nm) and the temperature. Here’s an example for D-(+)-glucose.

7. Calculation of Specific Rotation: A Sample Problem
Most problems involving specific rotation will ultimately just require a bit of high school algebra. “Plug and chug,” so to speak. Here’s an example:
A sample containing a single enantiomer of fluoxetine (Prozac) is placed in a polarimeter. The observed rotation is 9.06° clockwise. The sample was made by dissolving 1.24 g of fluoxetine in a solution with a total volume of 2.62 mL. The light source was a sodium D line and the temperature was 25° C. The length of the sample tube was 1.25 dm.
You can solve this problem with the following steps.
[α] = [+ 9.06° ] / [(1.24 g/ 2.62 mL)(1.25 dm)]
[α] = +15.3°
Note that we usually just report this number in degrees, although the actual units are degrees cm2 g-1
8. Conclusion: Optical Rotation And Specific Rotation
This post briefly covered some of the main details of optical rotation and specific rotation. In the next post, we’ll explore the relationship between specific rotation and a concept called “enantiomeric excess”.
Questions or comments about this post? Leave one below!
Thanks again to Matt for helping with this post. Hire Matt as your tutor!
Notes
Note 1. Thank whatever god you wish to pray to that Fischer guessed correctly, because the old chemical literature would be a bloody nightmare to navigate if he had guessed wrong.
Note 2. Material in this paragraph was adapted from this excellent historical article by crystallographer Howard Flack – highly recommended. In particular, I love Pasteur’s account of demonstrating the resolution of racemic acid to his mentor, Biot.
From John Herschel’s 1822 publication (online here) – a student of organic chemistry might find the following description familiar…
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
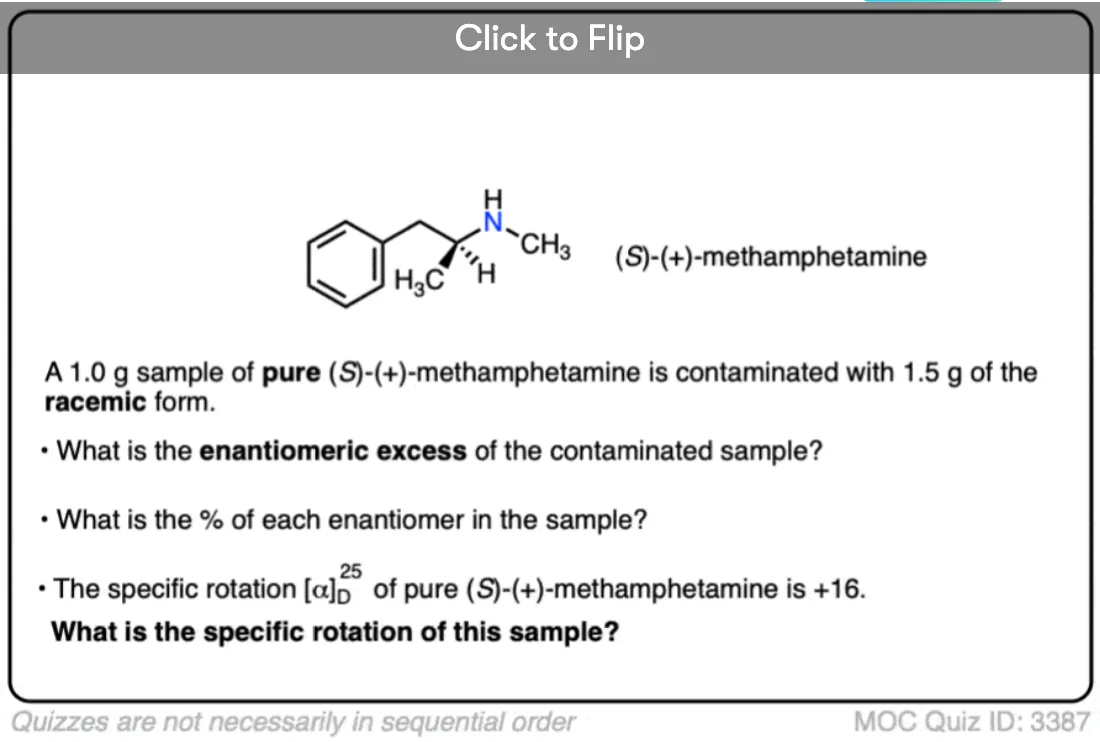
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Vogel’s Textbook of Practical Organic Chemistry, 5th Edition, rev. by Brian S. Furniss et. al. 1989. John Wiley & Sons, NY, 1991. Page 248.
- The resolution of racemic acid: A classic stereochemical experiment for the undergraduate laboratory
George B. Kauffman and Robin D. Myers
Journal of Chemical Education 1975, 52 (12), 777
DOI: 1021/ed052p777
This paper goes into detail on Pasteur’s life and background and also shows how Pasteur’s experiment can be repeated by undergraduates! - The discovery of biological enantioselectivity: Louis Pasteur and the fermentation of tartaric acid, 1857—A review and analysis 150 yr later
Joseph Gal
Chirality 2008, 20 (1), 5-19
DOI: 1002/chir.20494
Another historical review discussion Pasteur’s life and his work on the fundamentals of chirality in organic chemistry. - A model for optical rotation
L. L. Jones and Henry Eyring
Journal of Chemical Education 1961, 38 (12), 601
DOI: 10.1021/ed038p601
For those interested in the physics or theoretical underpinnings of the phenomenon of optical rotation, this paper by Prof. Henry Eyring (the same of Eyring equation fame) is a very good read. - Optical rotation
O. M. Evans and H. R. Tietze
Journal of Chemical Education 1964, 41 (12), A973
DOI: 10.1021/ed041pA973.2
Simple procedure for measuring the optical rotation of sucrose. - Optical activity in small molecules, nonenantiomorphous crystals, and nematic liquid crystals
Kenneth O’Loane
Chemical Reviews 1980, 80 (1), 41-61
DOI: 10.1021/cr60323a003
The first of this review is relevant for this topic, and covers Pasteur’s experiment as well as the various forms of chirality in organic small molecules. - IUPAC tentative rules for the nomenclature of organic chemistry. Section E. Fundamental stereochemistry
IUPAC-IUB Comm. on Biochem. Nomenclature
The Journal of Organic Chemistry 1970, 35 (9), 2849-2867
DOI: 1021/jo00834a001
These can be considered as the official IUPAC rules for nomenclature! - Specification of Molecular Chirality
R. S. Cahn, Sir Christopher Ingold, V. Prelog
Angew. Chem. Int. Ed. 1966, 5 (4), 385-415
DOI: 10.1002/anie.196603851
This is not the first paper on the topic by the authors (see Refs. 4 and 5), but it is a major publication and an attempt to consolidate all the information on chirality in a single place. This paper discusses the various types of chirality possible in chemistry (not just at tetrahedral carbons!) and how to assign chirality unambiguously. - Basic Principles of the CIP‐System and Proposals for a Revision
Dr. Vladlmir Prelog and Prof. Dr. Günter Helmchen
Angew. Chem. Int. Ed. 1982, 21 (8), 567-583
DOI: 10.1002/anie.198205671
An update to Ref. #1, which addresses a lot of edge cases that may come up in complex stereochemical assignments. - CHIRALITY IN CHEMISTRY
Vladimir Prelog
Nobel Lecture, 1975
https://www.nobelprize.org/prizes/chemistry/1975/prelog/lecture/
Prelog’s Nobel Lecture. Nobel Lectures are fascinating to read as they give insight into the life of scientists and the path to discovery, which is rarely linear. - “Absolutely” simple stereochemistry
Philip S. Beauchamp
Journal of Chemical Education 1984, 61 (8), 666
DOI: 10.1021/ed061p666
This paper describes a simple method for determining stereochemistry of tetrahedral carbons using the hands, suitable for undergraduate students of organic chemistry. - A simple hand method for Cahn-Ingold-Prelog assignment of R and S configuration to chiral carbons
Martin P. Aalund and James A. Pincock
Journal of Chemical Education 1986, 63 (7), 600
DOI: 10.1021/ed063p600
A follow-up to the previous paper (Ref #4), but sadly it is incomplete! - A Web-Based Stereochemistry Tool to Improve Students’ Ability to Draw Newman Projections and Chair Conformations and Assign R/S Labels
Nimesh Mistry, Ravi Singh, and Jamie Ridley
Journal of Chemical Education 2020, 97 (4), 1157-1161
DOI: 10.1021/acs.jchemed.9b00688
This paper discusses a web-based tool that helps students with visualization of chiral compounds and assignment of stereochemistry as per the Cahn-Ingold-Prelog (CIL) rules. See ref. 34 in the paper for the link.
this was so helpful. now wondering why i didnt just come here in the first place i could have gotten an a plus for my organic chemistry exam
Plz give one suggestion to me
Malic acid , specific optical rotation (-0.1 to +0.1) why
and what are meaning of (-0.1 to +0.1) in subjected specific optical rotation
plz suggeste to me
with regards
Ajay k. Maurya
Hi! I’m still confused with one thing. Are diastereomers always optically active? When is it not and when is it active?
Whether a molecule is optically active is an independent property relative to whether or not it has a diastereomer.
For example cis and trans 2-butene are diastereomers, but the molecules are achiral, because each molecule has a plane of symmetry (in the plane of the page).
That’s the key thing when determining whether or not a molecule is capable of being optically active. Does it have a plane of symmetry?
Generally, a molecule with only one chiral center will not have a plane of symmetry, and therefore can be optically active.
Most molecules with two chiral centers also lack a plane of symmetry, and are therefore optically active, but you need to *watch out* for meso compounds, because these are molecules which have 2 (or more) chiral centers but are arranged in such a way that the molecule lacks a plane of symmetry.
(Just one last point. Don’t forget that the terms “enanantiomers”, “diastereomers”, etc. are about relationships between two molecules, just like “brother”, “cousin”, “father” are relationships between people. It’s possible for a molecule to be both an enantiomer of one molecule and a diastereomer of another, just like it’s possible for a person to be both a brother to one person and a father to another. That’s why I say that physical properties like optical rotation are independent of those terms.)
we are getting the Specific optical rotation results exact double against the limit what are probable reason as we have cross checked all the parameters but unable to find out the root cause.
If you had a sample volume of 2.62 mL and the cell was 12.5 cm, the cross-sectional area of the tube would be about 0.2 cm^2 and its diameter would be about 5 mm. Do people often do polarimetry in NMR tubes?
Ha! That is a great point Dave!
The pure sample of the R isomer of a compound has [𝛼] = +5.756°. If you will prepare a
solution with 0.300 mol of the R isomer, and 0.700 mol of the S isomer, what will be your
prediction of the [𝛼] of the solution?
Was having a problem with this question in a textbook:
“If a solution of a compound has a rotation of +12, how could
you tell if this was actually +12, or really –348, or +372?”
For which the solution implies using [a] = a / cl & changing concentration should enable you to deduce the answer. But I’m not quite seeing how, would you be able to explain please?
Dilute it by half. You will either get +6 or +181.
Hi Dale,
I was wondering if there is a difference in specific rotation between anhydrous and monohydrous salt forms of a drug substance. If yes, how can this be calculated?
Do you have a specific example?
So from this (and my textbook) it seems there is no direct correlation between (S), (R) and optical rotation correct? I thought I had this all in hand but I got the following question incorrect on my exam “If (S)-2-ethyl-1-hexanol has a specific rotation of -20.05 degrees, what is the specific rotation of (R)-2-ethyl-1-hexanol?”. I don’t know what the correct answer would be in this case, can you help?
Hi Sarah – Between any random molecule with (S) and any random molecule with (R) you are right that there is no simple correlation to the direction of rotation of polarized light.
However these two molecules are not just any random molecule! The key part of this question is recognizing that (S)-2-ethyl-1-hexanol and (R)-2-ethyl-1-hexanol are *enantiomers*.
It is true that enantiomers rotate plane-polarized light to exactly equal and opposite extents. That’s why racemic mixtures don’t rotate polarized light – the rotations cancel out.
Therefore if (S) has a specific rotation of -20.05 degrees, the specific rotation of (R) will be +20.05 degrees.
I got the polarimeter reading as -0.039 for jasmine essential oil. so how do i calculate Optical rotation and specific rotation.
Essential oils are usually complex mixtures of terpenes. Are you dealing with a pure compound? If not, optical rotation is useless.
Am finding 15.31 after computing! Is there a mistake in your calculation?
Yes. Fixed. Thank you for spotting the error!
In the above-titled section on your website, you state “For example, for (S)-malic acid at a concentration of 5.5 g/ mL in the solvent pyridine….”. This concentration cannot be correct. If you combine 5.5 g malic acid (or almost any solid) with only 1 mL of pyridine (or almost any solvent), you will get a sticky mess. Furthermore, 5.5 g of malic acid is 0.041 mol of acid while 1 mL of pyridine is 0.0124 mol of base, so 0.0124 mol pyridine will simply react with 0.0124 mol of malic acid, make 0.0124 mol of a salt and probably just solidify in the rest of the malic acid. Shoud the concentration be 5.5 g/L or 0.55 g/mL?
Shouldn’t it be 5.5 g malic acid/100 mL pyridine? I know for pure liquids the unit is g/mL while for solutions, it’s g/100 mL
That is correct. I’ve updated the article to reflect this. Thank you very much.
Hey there!
I did an experiment where i increased the concentration of sugar in a sugar-water solution and calculated the observed angles. I used a different wavelength than D and the temp was about 30 deg C and a pathlength of about 25 cm. I graphed concentration vs angle to find a linear relationship as expected. However, i checked and the specific rotation of sugar (sucrose) is +66. However, when i put my observed angles and concentrations into the equation (about 11 deg at 25g/100mL water), my specific rotation was much lower. Is this normal? Also how do i use the slope of my graph to find out the specific rotation? Do i have to put the individual points into the equation one by one or can is there a way where I can just use the slope? What can I do with this graph?
if single bond rotates, why glucose does not get converted into mannose or other structural isomers??
Because glucose and mannose have different *configurations*. Rotating a single bond does not change the configuration of glucose, in the same way that no amount of wiggling your left hand will turn it into your right hand.
In this case (for historical reasons), c = 1.0 translates to 10 mg of sample in 1.0 ml of solvent.
I believe in the sample problem you have made the incorrect calculation Note the added parentheses in the equation. It should be:
[α] = [+ 9.06° ] / [(1.24 g/ 2.62 mL) x 1.25 dm]
[α] = +15.3
That is correct. My mistake. I greatly appreciate the correction Dale.
“For example, for (S)-malic acid at a concentration of 5.5 g/ mL in the solvent pyridine at 20°C at a wavelength of 589 nm, the specific rotation is –27° . We can describe this in shorthand as [α]20D –27° (c = 5.5, pyridine)”
How does one get 5.5 grams into one mL ? So what does c=5.5 actually mean?
Since there is no direct correlation between R and S and the optical rotation of a chiral compound.
To check the chirality of a chiral compound, the specific rotation is measured on the polarimeter, using a specified solvent. Then can the specific rotation result be used for confirmation of the R or S configuration of the molecule?
5.5 is g/100mL
Yes, thank you.