Reactions of Aromatic Molecules
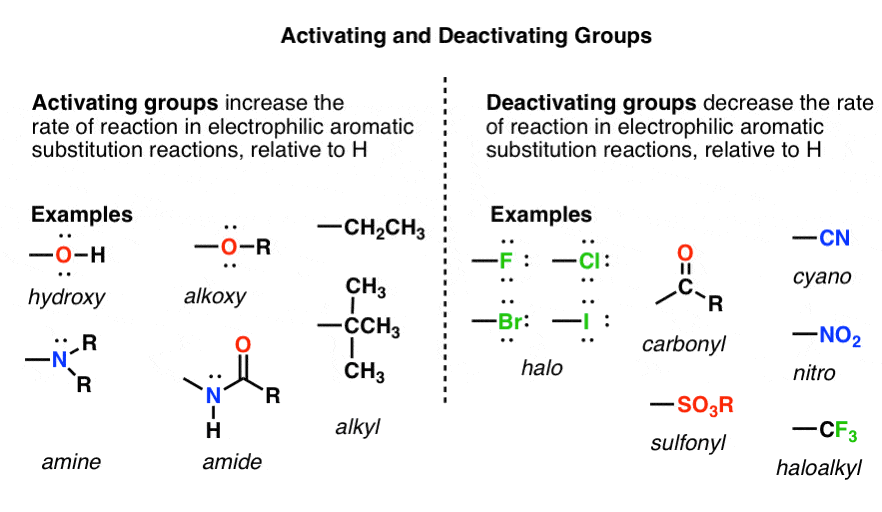
Activating and Deactivating Groups In Electrophilic Aromatic Substitution
Last updated: February 7th, 2025 |
Activating and Deactivating Groups in Electrophilic Aromatic Substitution
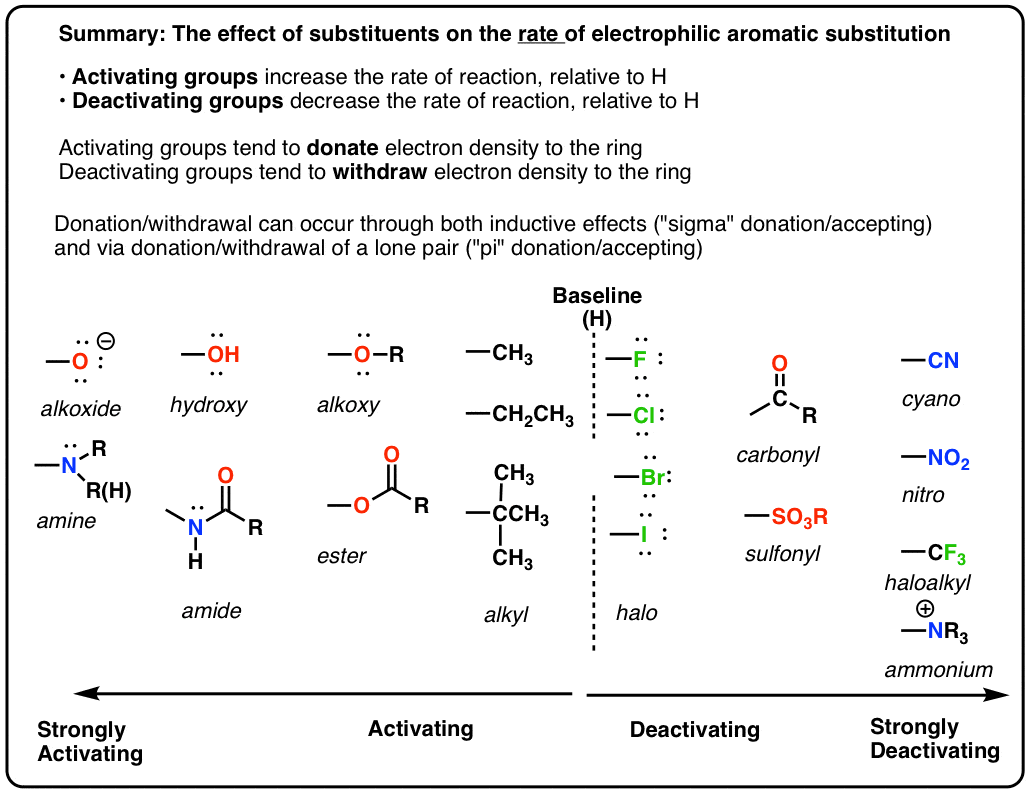
- The rate of electrophilic aromatic substitution (EAS) reactions is greatly affected by the groups attached to the ring. The more electron-rich the aromatic ring, the faster the reaction
- Groups that can donate electron density to the ring make EAS reactions faster.
- If a substituent increases the rate of reaction relative to H it is called activating. If it decreases the rate relative to H it is called deactivating. (These rates need to be measured by experiment).
- Important! Groups like OR and NR2 that seem like they should be deactivating because of their electronegativity are actually activating since they can donate a lone pair of electrons into the ring through resonance.
There’s a lot to this post, so here’s a quick index:
Table of Contents
- Activating And Deactivating Groups
- Measuring Reaction Rates Can Provide Insight Into The Mechanism
- “Activating” and “Deactivating” Groups – A Definition
- “Sigma” (σ) donors and acceptors (otherwise known as “inductive effects”)
- Pi ( π) Donors and Acceptors (often just called “Resonance”)
- Oxygen And Nitrogens Containing Lone Pairs Are Highly Activating When Bonded Directly To The Ring
- Halogens (F, Cl, Br, I) Are Deactivating
- Pi Acceptor Groups Are Strongly Deactivating
- A Table of Activating and Deactivating Groups
- Summary: What Does This Tell Us About The Mechanism Of Electrophilic Aromatic Substitution?
- Quiz Yourself!
- Notes
- (Advanced) References and Further Reading
1. Activating And Deactivating Groups
Last post in this series we introduced electrophilic aromatic substitution. Here’s the general case:
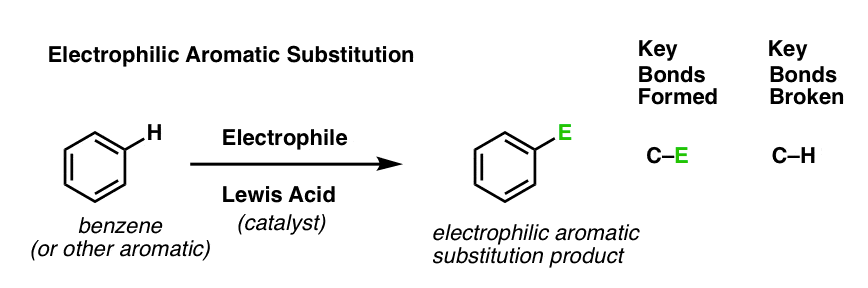
Why is this a substitution reaction, you ask? Because we’re forming and breaking a bond on the same carbon. We form C–E (where “E” is a generic term for “electrophilic atom”) and we break C–H.
[As for the specific identity of “E”, we mentioned six key electrophilic aromatic substitution reactions in the last post (bromination, chlorination, nitration, sulfonylation, Friedel-Crafts alkylation and Friedel-Crafts acylation) that we’ll eventually dig into in detail. But not yet. ]
So if that’s the summary of what happens, the next obvious question is: how does it happen?
In other words, what’s the mechanism?
Obligatory pre-mechanism speech: You can’t determine the mechanism of a chemical reaction merely through logical deduction from first principles. Sure, you can make guesses – even good ones! But the ultimate test of a mechanistic hypothesis is how well it fits with experiment, and that typically involves a lot of lab work. What you’re taught in an introductory course is the tippy-topmost layer of snow on the iceberg. We give you the best answer, and in retrospect it looks obvious. What you don’t see is all the failure, wrong turns, and false hypotheses that happened along the path towards determining the correct mechanism. However, the mechanisms of these reactions that you will learn about weren’t obvious to most of their discoverers, who were among the brightest and best chemists of their time. Remember that.
2. Measuring Reaction Rates Can Provide Insight Into The Mechanism
As far as determining mechanisms is concerned, one of the best tools we have in our experimental arsenal is the ability to measure reaction rates.
By measuring the effect that slight tweaks in the experimental conditions (e.g. structure of reactant, temperature, solvent) have upon the rate, we can gather useful insights about how a reaction operates “under the hood”.
Of the parameters mentioned above, changing the substrate (reactant) is probably the most powerful way to probe a mechanism, because it allows you to tune how electron-rich (nucleophilic) or electron-poor (electrophilic) it is.
Let me show you what I mean.
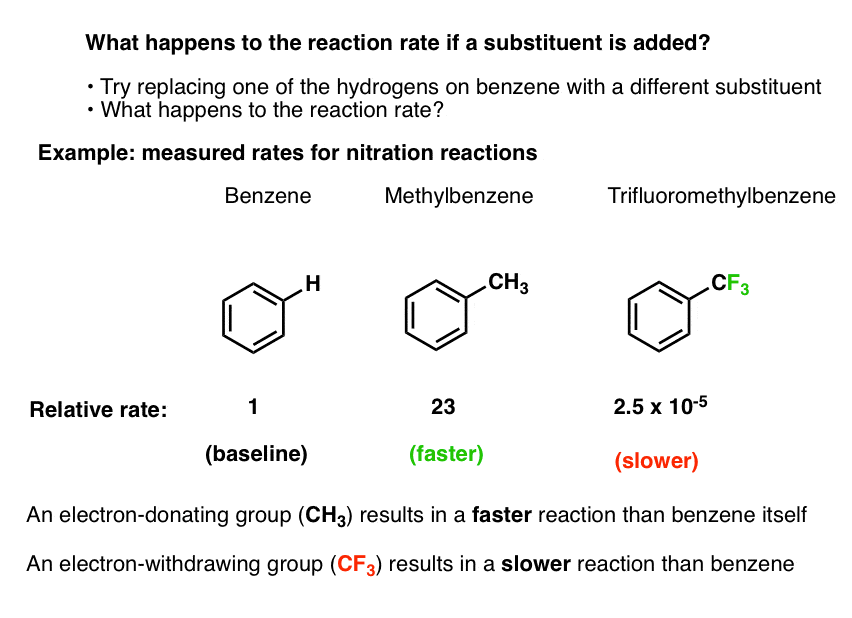
Let’s arbitrarily pick one electrophilic aromatic substitution reaction: nitration.
- We know that by adding nitric acid and H2SO4, benzene can undergo nitration to form nitrobenzene (break C-H, form C-NO2)
- We can even measure the rate of this reaction at a given temperature, concentration, and solvent.
- Using the exact same experimental conditions we can then measure the rate of the reaction when toluene (methylbenzene, C6H5CH3) is used as the substrate instead of benzene.
- The nitration of toluene is 23 times faster than it is for benzene. [Ref 1]
- Using the exact same experimental conditions, we can also use trifluoromethylbenzene (C6H5CF3) as the substrate, and measure the reaction rate.
- The nitration of trifluoromethylbenzene is 40,000 times slower than it is for benzene (2.5 × 10-5).
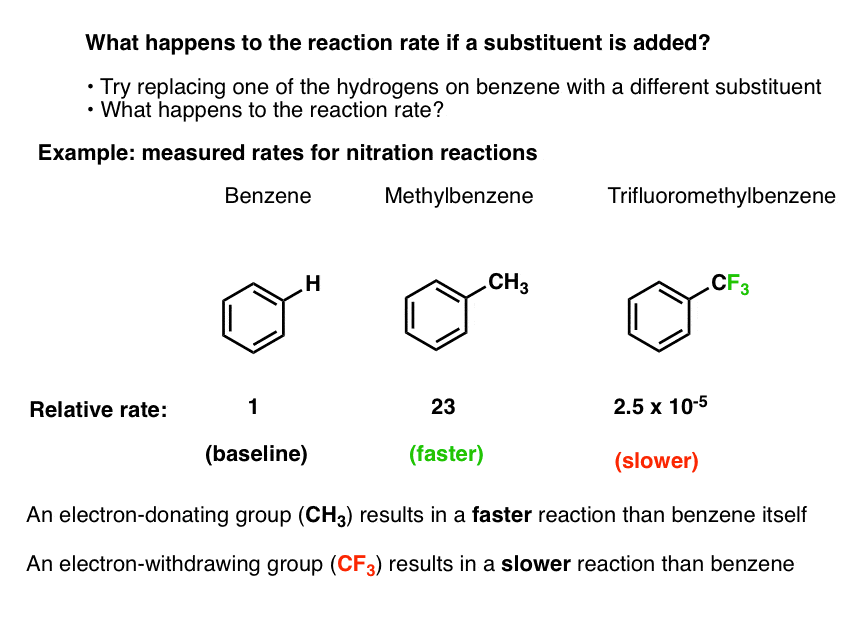
Bottom line: if we swap a hydrogen on benzene for a methyl group, the reaction is faster. If we swap a hydrogen for a trifluoromethyl group, the reaction is slower.
{kind=link}
This pattern turns out to be general for other electrophilic aromatic substitution reactions as well (chlorination, bromination, Friedel-Crafts, and others).
3. “Activating” and “Deactivating” Groups – A Definition
Let’s call a group activating that increases the rate of an electrophilic aromatic substitution reaction, relative to hydrogen. As we just saw, CH3 is a perfect example of an activating group; when we substitute a hydrogen on benzene for CH3, the rate of nitration is increased.
A deactivating group, on the other hand, decreases the rate of an electrophilic aromatic substitution reaction, relative to hydrogen. The trifluoromethyl group, CF3 , drastically decreases the rate of nitration when substituted for a hydrogen on benzene.
This definition is ultimately based on experimental reaction rate data. It doesn’t tell us why each group accelerates or decreases the rate. “Activating” and “deactivating” just refers to the effect of each substituent on the rate, relative to H.
OK then. So why might CH3 increase the rate of reaction, and CF3 decrease it?
4. “Sigma” (σ) donors and acceptors (otherwise known as “inductive effects”)
Let’s quickly think back to what we know about alkyl groups (such as CH3) and haloalkyl groups (such as CF3), and try to address this question.
In CH3, the carbon atom is more electronegative (2.5) than hydrogen (2.2). This means that the carbon attracts a bit more than an equal share of electron-density from the covalent bond with H, resulting in a partial negative charge (δ–) on carbon and a partial positive charge (δ+) on hydrogen. This partial negative charge is then available to be donated to an adjacent atom. Hence, we tend to think of CH3 as an electron-rich species; an electron–donor.

In CF3 the electrons are pulled in the opposite direction. Three highly electronegative (4.0) fluorine atoms pull electron density away from the carbon atom (2.5), resulting in a partial positive charge (δ+) on carbon. Rather than donate electron density, the carbon tends to accept (pull away) electron density from adjacent atoms (this is the familiar inductive effect) We generally consider CF3 to be an electron-poor species; an electron-acceptor.

Since these inductive effects operate solely through single bonds (“sigma”, or σ bonds) this behaviour is sometimes called “sigma donation” (as for CH3) or “sigma accepting” (for CF3).
So it seems like a good hypothesis that
- activating groups are electron-donating (relative to H), and
- deactivating groups are electron-withdrawing (relative to H)
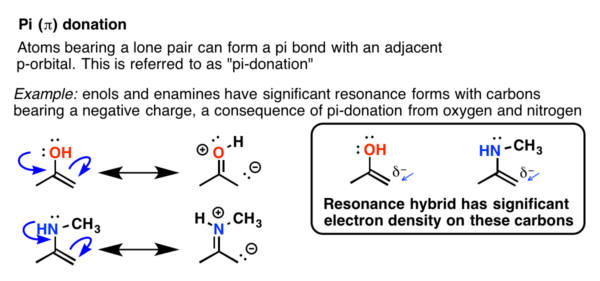
5. Pi ( π) Donors and Acceptors (otherwise known as “Resonance”)
Sigma donation and acceptance helps us to understand the effect of alkyl groups on electrophilic aromatic substitution. So what about other functional groups? What effect might, say, a hydroxyl group have on the rate of nitration?
Quiz time. Do you think –OH would be activating (increase the rate) or deactivating (decrease the rate) for electrophilic aromatic substitution (such as nitration)? Guessing is OK!

Based on what we just said, it’s fully understandable if you said, “deactivating”. After all, oxygen is highly electronegative (3.4) and through induction, pulls away electron density through the bond. In other words, it’s a sigma-acceptor.
The fact is, however, that OH greatly accelerates the rate, orders of magnitude more than CH3 does. In fact I couldn’t find good rate data comparing OH to CH3 because in the case of -OH, the reaction is what’s called, “diffusion controlled”. That roughly means, “as soon as the reactant comes in contact with the electrophile, a reaction occurs.” In other words, the –OH group is highly activating.
Clearly, something else must be going on here besides the inductive effect of oxygen!
6. Oxygen And Nitrogens Containing Lone Pairs Are Highly Activating When Bonded Directly To The Ring
As we saw in our chapter way back on resonance, hydroxyl groups are excellent pi donors. The lone pairs on the oxygen atom can form a pi bond with an adjacent atom containing an available p-orbital.
{kind=link}
This donation effect (or “resonance”) must outweigh electron-withdrawal via inductive effects, otherwise we’d observe that hydroxyl groups are deactivating.
The same is true for nitrogen groups with lone pairs, such as amines and amides (below).
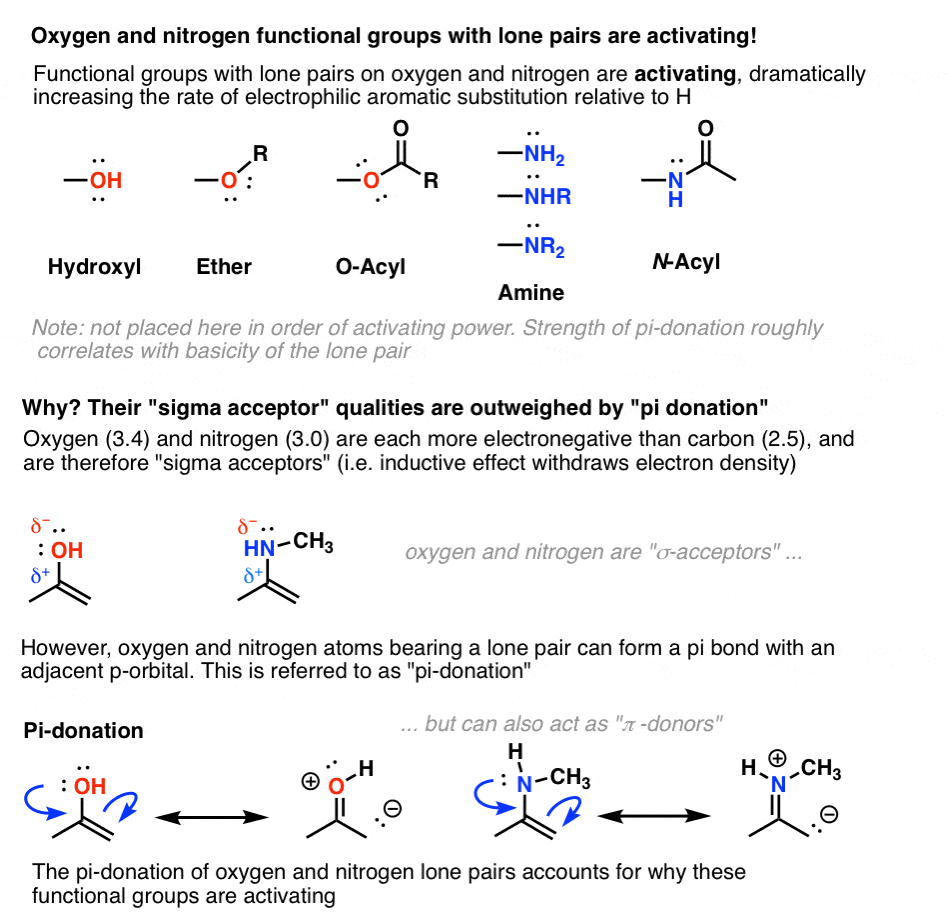
[One measure of the importance of pi-donation in the activating nature of amines is seen in their behavior under strongly acidic conditions. If the nitrogen lone pair is either protonated with strong acid or undergoes a substitution reaction to form NR3+ , pi-donation is impossible and the group becomes strongly deactivating (see table below). ]
7. Halogens (F, Cl, Br, I) Are Deactivating
Not all groups capable of pi donation are activating groups. For example, halogens (F, Cl, Br, I) tend to be deactivating. The rates of electrophilic aromatic substitution reactions on fluorobenzene, chlorobenzene, bromobenzene, and iodobenzene are all slower than they are for benzene itself. In these cases, inductive effects (“sigma accepting”) would appear to have a greater effect on the rate than any pi-donation from the lone pairs. [pi donation < sigma acceptance]. [Why?]
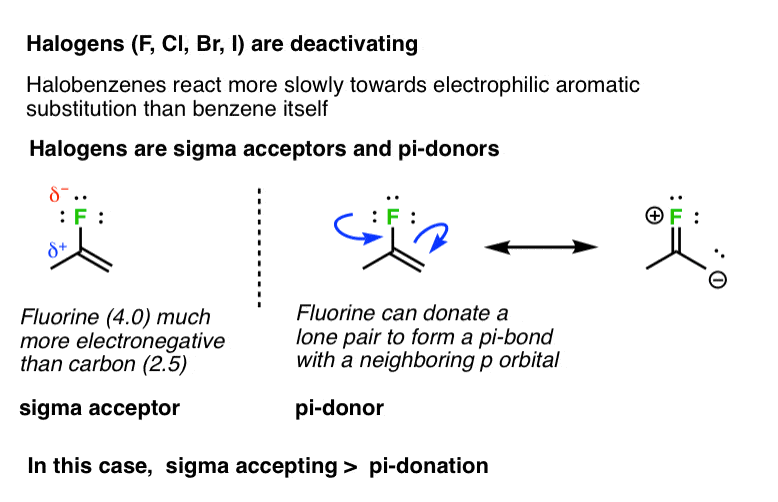
A good rule of thumb for pi-donation ability is the basicity of the lone pair. Amines tend to be better bases than oxygens, which are far better bases than halogens.
Alright. What if electrons flow in the opposite direction? Is there an opposite of “pi donor” ?
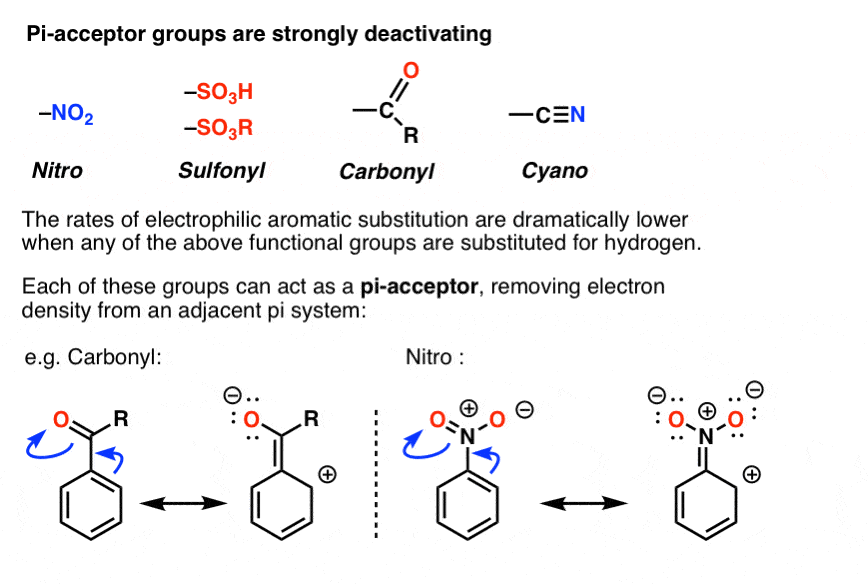
8. Pi Acceptor Groups Are Strongly Deactivating
Yes! As you may already know, the opposite of a “pi-donor” is a “pi acceptor”. Certain functional groups can accept, rather than donate, a pi bond from the ring, resulting in a new lone pair on a substituent atom. Examples are NO2, carbonyl groups (C=O), sulfonyl, cyano (CN) among others. These groups are universally deactivating, slowing the rate of electrophilic aromatic substitution.
In terms of resonance, one can draw a pi bond from the aromatic ring forming a pi bond with the atom bound to the ring, resulting in formation of a new lone pair on an electronegative atom on the substituent. Note how this results in a positive charge on the ring!
So how do we keep all of these factors straight?
This is an example of why I say that resonance is the most important key concept to review for Org 2. In the section on aromatic chemistry it comes back with a vengeance.
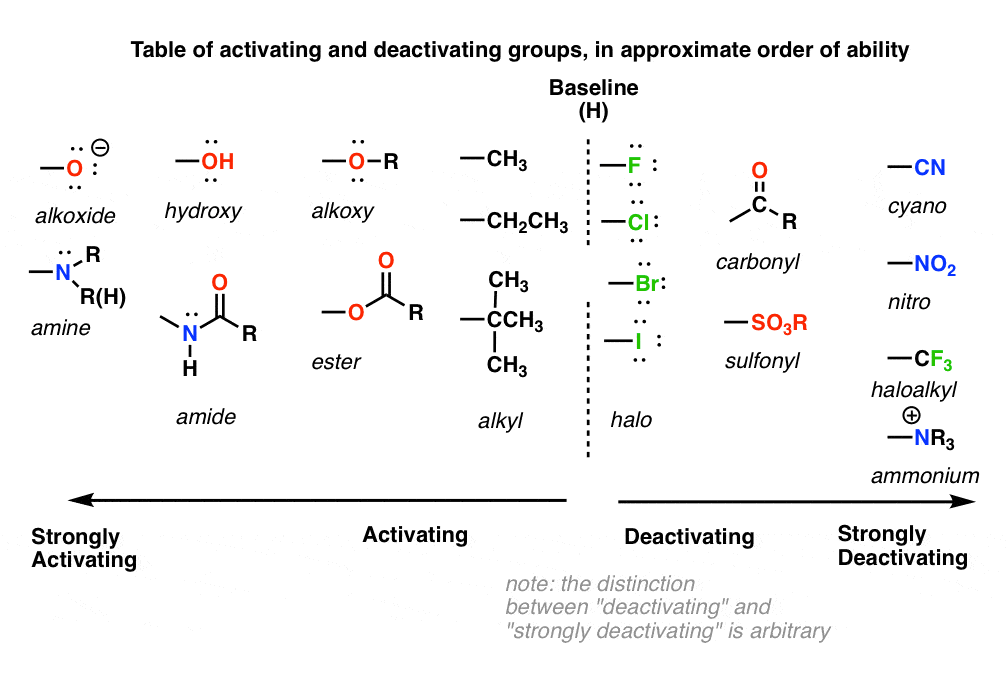
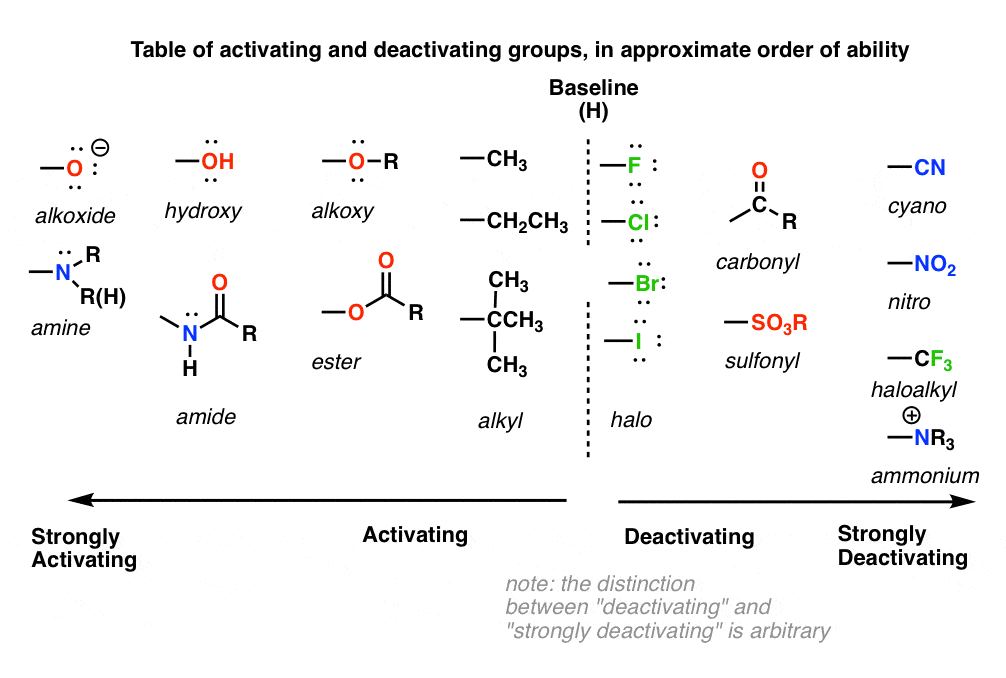
9. A Table of Activating and Deactivating Groups
Now seems like the right time to present a big table of activating and deactivating groups. It’s hard to rank exactly by power since the effect is averaged over several types of reactions.
Oh dear, this looks like a lot to remember. How to keep it all straight?
{kind=link}
I would suggest five main “buckets”, below:
- Nitrogen and oxygens with lone pairs – amines (NH2, NHR, NR2), phenol (OH) and its conjugate base O– are very strong activating groups due to pi-donation (resonance). Alkoxy, amide, ester groups less strongly activating.
- Alkyl Groups – (with no electron withdrawing groups). Moderately activating through inductive effect.
- Halogens – Moderately deactivating. Electron withdrawing (highly electronegative) nature outweighs donation of electron density through a lone pair.
- Atoms with pi-bonds to electronegative groups – Strongly deactivating. NO2, CN, SO3H, CHO, COR, COOH, COOR, CONH2. All pi-acceptors.
- Electron withdrawing groups with no pi bonds or lone pairs – Strongly deactivating. CF3, CCl3, and NR3(+). Pure inductive effect.
Once you remember the somewhat counterintuitive fact that O and N-bonded functional groups with lone pairs are activating, and halogens are deactivating, the rest is fairly straightforward.
One final word. Our table of “activating” and “deactivating” groups turns out to be a little bit like a pKa table. How? We can evaluate several factors that have an impact on pKa, but the ultimate test of which factor is more important is experimental measurement of an equilibrium constant. Likewise, with activating and deactivating groups, we can identify factors which may or may not make a certain group activating or deactivating, but in the end, its position on the chart comes down to experimental measurements of reaction rates.
 Click to Flip
Click to Flip
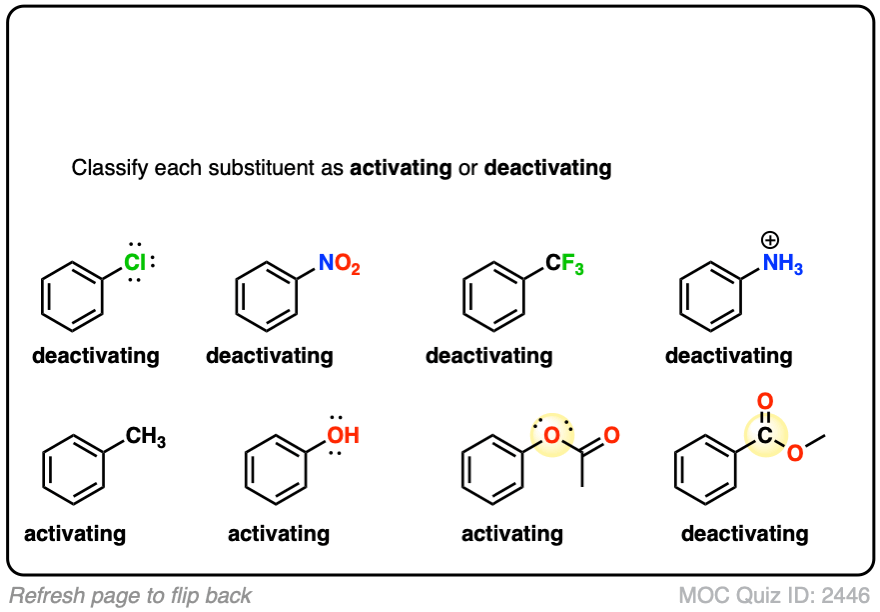
10. Summary: What Does This Tell Us About The Mechanism Of Electrophilic Aromatic Substitution?
OK. So what does all of this tell us?
Since the rate is so sensitive to whether the group is electron donating or electron withdrawing (“electronic effects”, as organic chemists might quickly summarize it) it would suggest that the rate determining step is the formation of a fairly unstable electron-poor species, such as a carbocation.
Recall CH3 and CF3. You may recall that the order of carbocation stability (tertiary > secondary > primary) is due to the fact that carbocations are stabilized by adjacent alkyl groups (such as CH3), and, conversely, are destabilized by adjacent electron withdrawing groups (like CF3).
Likewise, carbocations are stabilized by adjacent atoms that can donate lone pairs (e.g. O and N) through resonance, and destabilized by pi acceptors such as C=O, NO2, and so on.
A likely first step would be something like this:
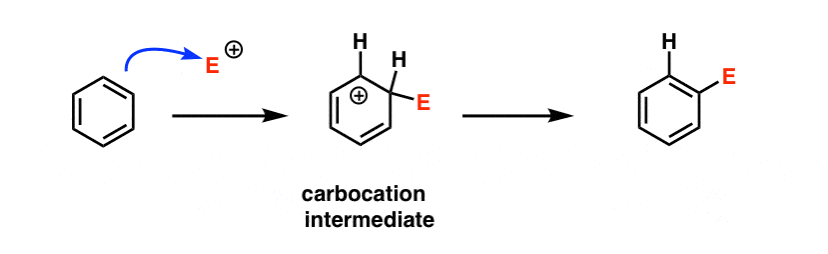
We’ll go into the full mechanism of electrophilic aromatic substitution in the next post, but will fill in additional detail in a bonus topic below.
Next Post: Electrophilic Aromatic Substitution: The Mechanism
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
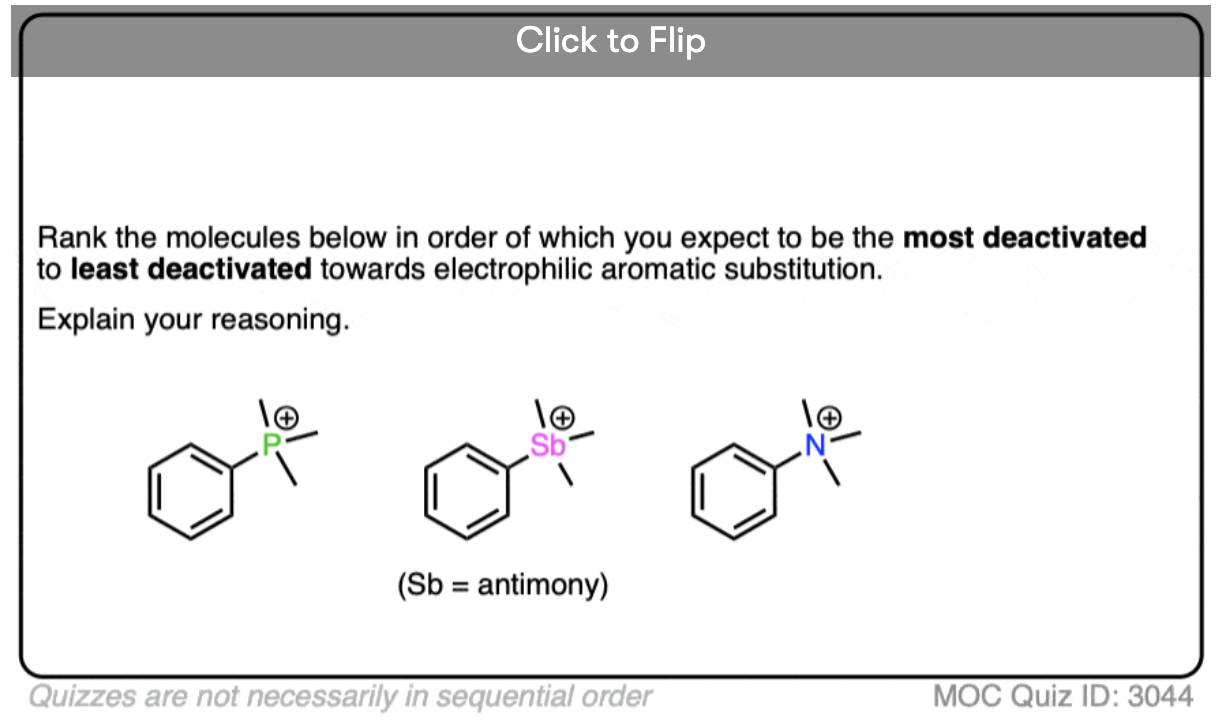
Become a MOC member to see the clickable quiz with answers on the back.
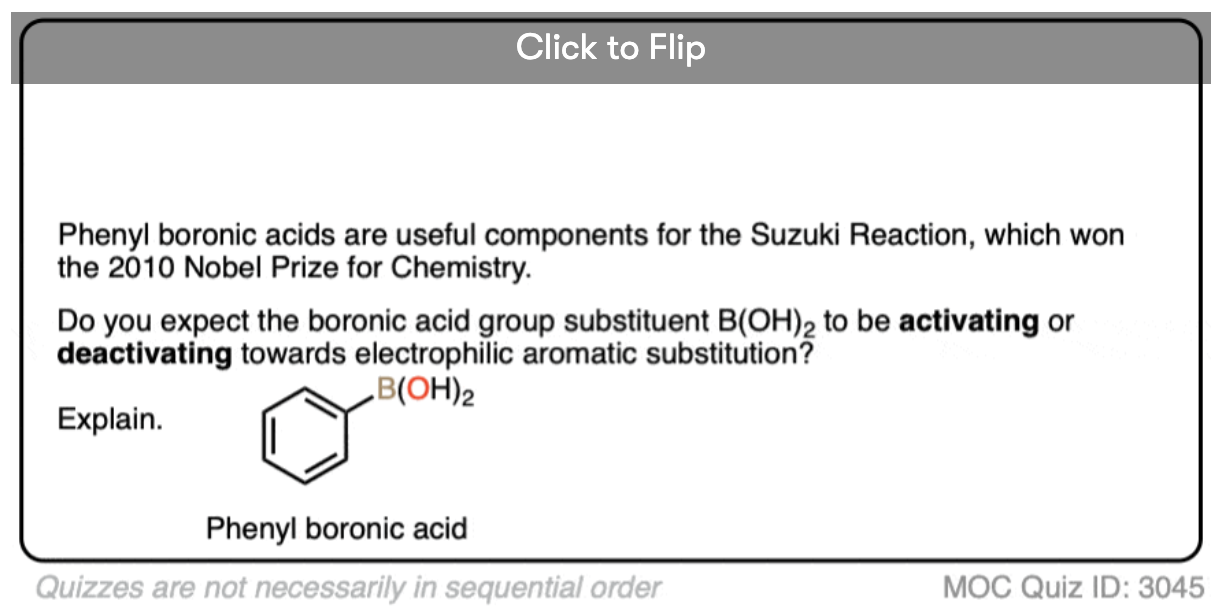
Become a MOC member to see the clickable quiz with answers on the back.
Notes
Possibly a useful reference sheet. Adapted from Ingold’s “Structure and Mechanism in Organic Chemistry”, 2nd ed.
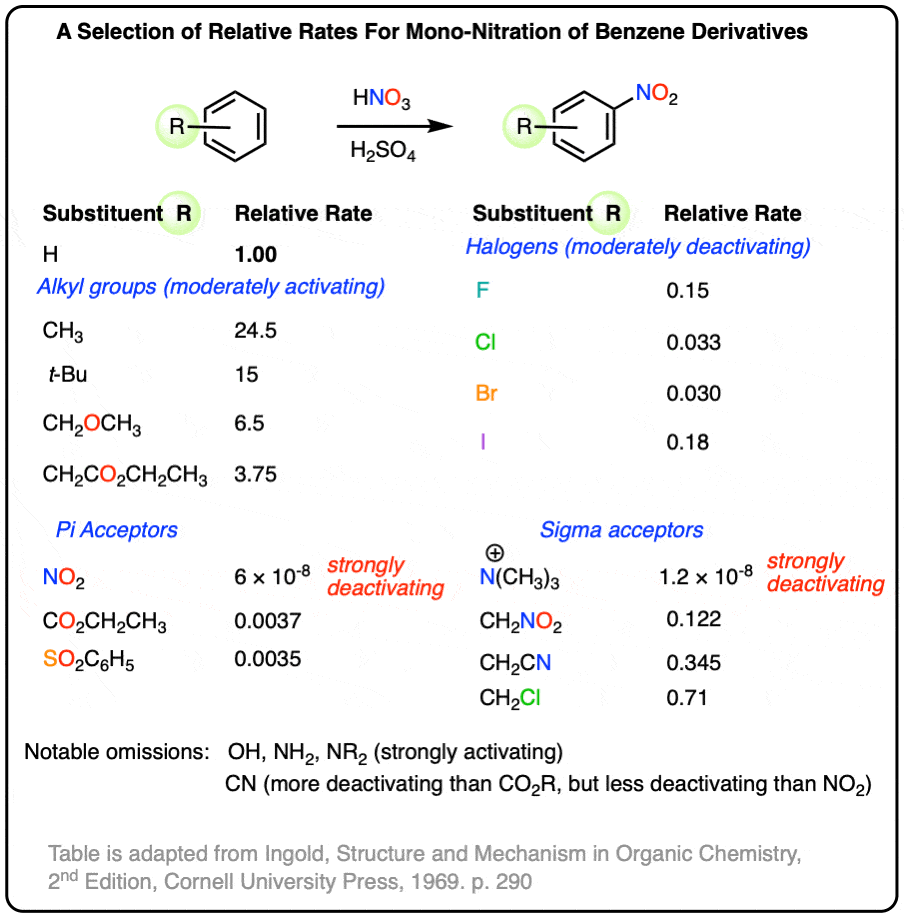
1. [Advanced] No deuterium isotope effect is observed in electrophilic aromatic substitution
In electrophilic aromatic substitution a C-H bond is broken. One way to probe the mechanisms of reactions that involve C-H bond cleavage is to use deuterium (D) labelling. In reactions where C-H bond breakage is a rate-determining step (e.g. E2 elimination) a C-H bond can break up to 6-7 times faster than a C-D bond. This is called a deuterium isotope effect and it is measurable.
Electrophilic aromatic substitution reactions have no significant deuterium isotope effects. [Note] This strongly suggests that C-H bond breakage is not the rate determining step.
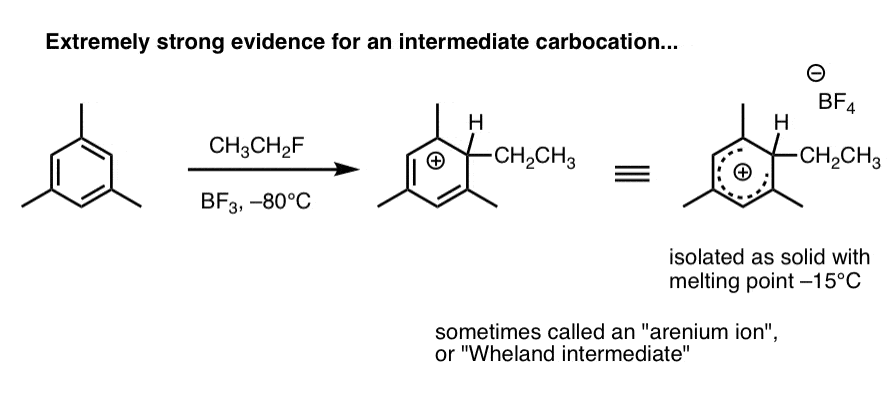
2. Carbocation intermediates have been isolated that strongly support the proposed mechanism
Here’s a species that’s been observed when 1,3,5-trimethylbenzene (mesitylene) is treated with ethyl fluoride and boron trifluoride at –80°C (this is a Friedel-Crafts alkylation reaction, by the way).
The carbocation intermediate (called an “arenium ion” or “Wheland intermediate” was isolated as a white solid with melting point –15°C, and analyzed by NMR spectroscopy.
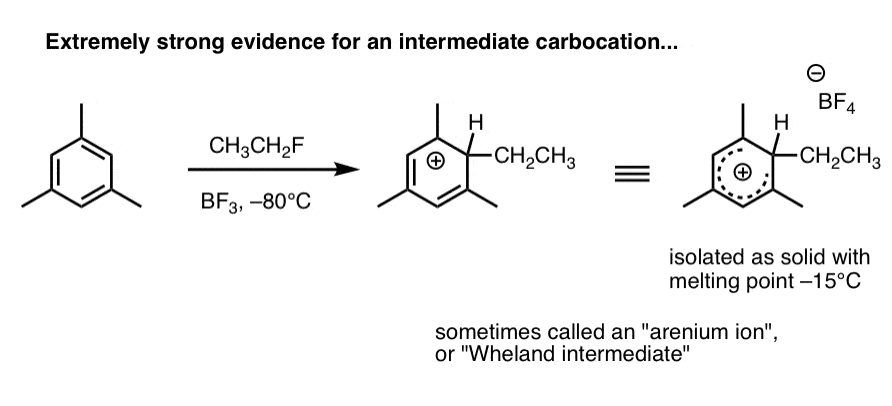
As Eric Jacobsen might say: “mechanisms can never be proven, but….” . (this pretty much seals the deal). We’ll go into in more detail in the next post.
{kind=link}
Note 1. Reference: March, Advanced Organic Chemistry 5th ed, page 692.
Note 2. Why? Interestingly, fluorine is the most activating of the halogens. The reason is likely that the overlap of the lone pair in the fluorine 2p orbital with the p orbital on carbon is much better (resulting in a stronger pi-bond) than is donation with the 3p (and higher) p orbitals of chlorine, bromine, and iodine.
Note 3. Actually a white lie; some electrophilic aromatic substitution reactions do have very small deuterium isotope effects, but we’re not touching that topic, nosiree. [partitioning effects, see March’s Advanced Organic Chemistry, 5th ed., p. 679]
(Advanced) References and Further Reading
As mentioned, this topic is useful for all types of EAS reactions – Friedel-Crafts alkylation/acylation, halogenation, nitration, etc.
- —The chlorination of anilides. The directing influence of the acylamido-group
Kennedy Joseph Previté Orton and Alan Edwin Bradfield
J. Chem. Soc. 1927, 986-997
DOI: 10.1039/JR9270000986
An early paper discussing the ortho/para product distribution obtained by electrophilic chlorination of anilides (generally 65% para/35% ortho). Unfortunately this paper does not have data comparing the rate of chlorination to benzene. - Kinetics and mechanism of some electrophilic benzene substitution reactions
Alan E. Bradfield and Brynmor Jones
Faraday Soc. 1941, 37, 726-743
DOI: 10.1039/TF9413700726
Table I in this paper contains partial rate factors for nitration of benzene and related compounds. Chlorobenzene and bromobenzene are around 1-3% as reactive as benzene, whereas ethyl benzoate is significantly deactivated – it is around 0.1-0.2% as reactive as benzene! Toluene is 40-50 times as reactive as benzene. - The kinetics of aromatic halogen substitution. Part IX. Relative reactivities of monosubstituted benzenes
P. W. Robertson, P. B. D. de la Mare, and B. E. Swedlund
J. Chem. Soc. 1953, 782-788
DOI: 10.1039/JR9530000782
Pg. 783 in this paper contains data for reaction rates of halogenation of various benzene derivatives. This spans the gamut of extreme activating substituents (N,N-dimethylaniline is 1018 times more reactive than benzene!) and deactivating substituents (nitrobenzene is 10-6 times less reactive than benzene). - The influence of the methoxyl group in aromatic substitution
P. B. D. de la Mare and C. A. Vernon
J. Chem. Soc. 1951, 1764-1767
DOI: 10.1039/JR9510001764
This paper examines the effect of -OMe in electrophilic aromatic substitution (e.g. anisole and related compounds vs. benzene). Overall, anisole is 108 times more reactive than benzene, and as a result, o/p selectivity in reactions is very low. - Rates of Bromination of Anisole and Certain Derivatives. Partial Rate Factors for the Bromination Reaction. The Application of the Selectivity Relationship to the Substitution Reactions of Anisole
Leon M. Stock and Herbert C. Brown
Journal of the American Chemical Society 1960, 82 (8), 1942-1947
DOI: 1021/ja01493a026
This paper is a more rigorous study of the bromination of anisole by Nobel Laureate Prof. H. C. Brown. The o/p selectivity of anisole is actually rather high – bromination gives 1.6% o– and 98.4% p-bromoanisole. The relative reaction of anisole:benzene is also measured to be 1.79 x 109:1.00. This paper also shows that s+ values (electrophilic Hammett constants) measured this way are comparable to Hammett values measured through other methods, and that the Hammett values also provide a good measure of how a substituent will effect EAS reactions as well. - —Influence of directing groups on nuclear reactivity in oriented aromatic substitutions. Part II. Nitration of toluene
Christopher Kelk Ingold, Arthur Lapworth, Eugene Rothstein, and Denis Ward
J. Chem. Soc. 1931, 1959-1982
DOI: 10.1039/JR9310001959
This is the first paper to introduce the term ‘partial rate factor’ (usually denoted by fp, fo, fm) to denote the amount by which a specific position on a substituted benzene may be more or less reactive compared to benzene. Table IV shows in this paper that toluene can be anywhere from 1.2 – 10 times more reactive than benzene. - Effects of Alkyl Groups in Electrophilic Additions and Substitutions
COHN, H., HUGHES, E., JONES, M. and PEELING, M. G.
Nature 1952, 169, 291
DOI: 1038/169291a0
This paper has data comparing the nitration of t-butylbenzene and toluene. T-butylbenzene is much more p-directing than toluene (79.5% para for t-butylbenzene vs. 40% para for toluene), which is likely due to sterics (ortho approach is blocked by the bulkier t-butyl group). - The transmission of polar effects through aromatic systems. Part II. The nitration of benzyl derivatives
J. R. Knowles and R. O. C. Norman
J. Chem. Soc. 1961, 2938-2947
DOI: 10.1039/JR9610002938
J. R. Knowles went on to become a Professor at Harvard, specializing in enzymology. The knowledge of kinetics that one gets from doing physical organic chemistry is applicable in a wide variety of areas! In this paper, Table 2 is interesting, and shows that the empirical reactivity difference between toluene and benzene is 25x, which is what is commonly found in textbooks today. T-butylbenzene is less reactive than toluene, but still 15x more reactive than benzene. - Influence of directing groups on nuclear reactivity in oriented aromatic substitutions. Part IV. Nitration of the halogenobenzenes
Marjorie L. Bird and Christopher K. Ingold
J. Chem. Soc. 1938, 918
DOI: 10.1039/JR9380000918
Table I in this paper shows that overall, chlorobenzene and bromobenzene are around 2-3% as reactive as benzene towards nitration under a wide variety of conditions. - Some aspects of the nitration of the mononitrotoluenes
J. G. Tillett
J. Chem. Soc. 1962, 5142-5148
DOI: 10.1039/JR9620005142
Pg. 5148 in this paper shows that in nitrotoluenes, the deactivating nature of nitro wins out over the activating nature of the methyl group. Interestingly, in m-nitrotoluene, the meta position to the nitro group is less reactive than the other positions, due to resonance effects. Note that these compounds are also precursors to the common explosive TNT! - Substituent effects of positive poles in aromatic substitution. Part I. The nitration of the anilinium ion in 90—100% sulphuric acid
Madeline Brickman and J. H. Ridd
J. Chem. Soc. 1965, 6845-6851
DOI: 10.1039/JR9650006845 - Substituent effects of positive poles in aromatic substitution. Part II. The nitration of N-methylated anilinium ions
Madeline Brickman, J. H. P. Utley, and J. H. Ridd
J. Chem. Soc. 1965, 6851-6857
DOI: 10.1039/JR9650006851
In contrast to aniline, which is very reactive in EAS compared to benzene, the anilinium ion (which is easily formed in acidic media) is deactivated. As the acidity of the medium increases, the amount of meta product obtained from nitration of aniline increases, indicating that the reaction is proceeding via the anilinium ion (PhNH3+). Reaction rates also decrease with increasing acidity, as the amount of free aniline available in the reaction gets lower and lower. - Aromatic substitution. 53. Electrophilic nitration, halogenation, acylation, and alkylation of (.alpha.,.alpha.,.alpha.-trifluoromethoxy)benzene
George A. Olah, Takehiko Yamato, Toshihiko Hashimoto, Joseph G. Shih, Nirupam Trivedi, Brij P. Singh, Marc Piteau, and Judith A. Olah
Journal of the American Chemical Society 1987, 109 (12), 3708-3713
DOI: 10.1021/ja00246a030
The -OCF3 substituent is not commonly encountered in undergraduate chemistry courses, but is used in medicinal chemistry. This paper by Nobel Laureate Prof. George A. Olah and his wife Judith, covers the directing effects and reactivity of PhOCF3 in a variety of EAS reactions. Overall, PhOCF3 is around 3-10% as reactive as benzene in EAS (see Tables VI-VIII). - A Quantum Mechanical Investigation of the Orientation of Substituents in Aromatic Molecules
G. W. Wheland
Journal of the American Chemical Society 1942, 64 (4), 900-908
DOI: 10.1021/ja01256a047
This discusses the structure of the arenium ion that gets formed in EAS reactions, also known as the s-complex or Wheland intermediate, after the author here who first proposed it. - Isolation of the Stable Boron Trifluoride – Hydrogen Fluoride Complexes of the Methyl-benzenes ; the Onium Salt (or σ-Complex) Structure of the Friedel-Crafts Complexes
OLÁH, G., KUHN, S. & PAVLÁTH, A.
Nature 1956, 178, 693–694
DOI: 1038/178693b0 - The Benzotrifluoride–Nitryl Fluoride–Boron Trifluoride Complex
OLÁH, G., NOSZKÓ, L. & PAVLÁTH, A.
Nature 1957, 179, 146–147
DOI: 1038/179146b0 - Isolation of the Stable Boron Trifluoride-ethylfluoride and Boron Trifluoride-formylfluoride Complexes of the Methylbenzenes: Mechanism of the Friedel–Crafts Reactions
OLÁH, G., KUHN, S.
Nature 1956, 178, 1344–1345
DOI: 10.1038/1781344a0
These papers by Nobel Laureate Prof. G. A. Olah describe the isolation and characterization of the intermediate ions (‘Wheland intermediates’) from various electrophilic aromatic substitution reactions – alkylation, nitration, and even protonation (by HBF4!) - A Quantitative Treatment of Directive Effects in Aromatic Substitution
Leon M. Stock, Herbert C. Brown
Phys. Org. Chem. 1963, 1, 35-154
DOI: 10.1016/S0065-3160(08)60277-4
This is a very comprehensive review for its time, summarizing work on directing effects in EAS (e.g. determining which groups are o/p-directing vs. meta-directing, and to what extent they direct/deactivate). - Stable carbocations. CLXX. Ethylbenzenium ions and the heptaethylbenzenium ion
George A. Olah, Robert J. Spear, Guisseppe Messina, and Phillip W. Westerman
Journal of the American Chemical Society 1975, 97 (14), 4051-4055
DOI: 1021/ja00847a031
This paper discusses the characterization of benzenium ions, which are intermediates in EAS, and the characterization of the heptaethylbenzenium ion, which is a stable species because it lacks a proton and therefore eliminates with difficulty. - The Anomalous Reactivity of Fluorobenzene in Electrophilic Aromatic Substitution and Related Phenomena
Joel Rosenthal and David I. Schuster
Journal of Chemical Education 2003, 80 (6), 679
DOI: 1021/ed080p679
A very interesting paper, suitable for curious undergrads, and discusses something that most practicing organic chemists will know empirically – fluorobenzene is almost as reactive as benzene in EAS or Friedel-Crafts reactions, which is counterintuitive when one considers electronic effects.
I do not see addressed a substituent such as an alkene…specifically a c double bonded to another carbon from the benzene ring. Is this deactivating because of resonance? Or activating because resonance is unlikely given it produces an unstable carbon anion?
If the electrophile does *not* hit the double bond, you are correct that a normal alkene should be slightly activating since it can act as a pi donor.
HOWEVER, One problem is that alkenes will react more easily with electrophiles than will the aromatic ring, so they are often destroyed during attempted electrophilic aromatic substitution. Attempted chlorination of styrene, for example, will end up hitting the alkene.
Electrophilic aromatic substitution can work when the alkene is in conjugation with an electron withdrawing group such as an ester, which helps to make the alkene more electron-poor.
Is there a specific example you are thinking about?
Will p-methoxybenzenesulfonic acid undergo a Freidel Crafts reaction?
Thank you so much…this page is a concept clearer….all the explanation part done
you should rewrite the section on “Halides competing effects” on the wikipedia page for “electrophilic aromatic directing groups”
I will take that as a compliment, Steven. Thanks
I just found out this blog and it’s make chemistry so simple and easy thanks for such an amazing blog
Sir,ACTIVATING and DEACTIVATING groups are defined with RELATIVE TO HYDROGEN. What does RELATIVE TO HYDROGEN really mean??
Please help me to sort it out sir🙏🙏
Measure the rate of the reaction with benzene. Then see what happens to the rate when one of the hydrogens on benzene is replaced by a different substituent like Cl, CH3, NO2, etc.
Which is more reacting among NR2 NHR NH2 OH?
FAN SIR….! You must provide Hard copies of your precious material…I surely bet….In India atleast ,it is going to get much love…😍
This website is a game changer. Wonderful strait-forward explanations. Thank you.
you have a typo in the first paragraph: “Friedel-Crafts alkylation and Friedel-Crafts alkylation” twice, instead of Acylation.
Thank you – and thank you for the correction! James
Alkoxy (O-R) is a -I group…
But it is activating and Ortho para directing..
Pls explain..
Also, does it increase or decrease the acidity of benzene?
Hi James
Yes CH3 is activating. I had a look on the internet and noticed the electrostatic potential map of benzene and methylbenzene are virtually identical? Should’nt there be more intensity of red about the ring in MB ikndicating there is greater electron density?
If you have 2 Ep maps for benzene and MB that show a difference in electron density about the rings please forward on.
Why are they appearing the same? i.e based on EP maps the reactivity of benzene and MB should be similar with little difference in reactivity. Of course this is born out in the lab !
Chemistry Educator in New Zealand
Hi Ray – rather than look at the potential map I would see if you can find the calculated electron densities. There shouldn’t be a huge difference but it would be significant enough to account for the 10-100 fold increase in rate for alkylbenzenes versus benzene itself.
which is more deactivting group b/w CN and CF3
According to calculations,CN is a *slightly* stronger electron withdrawing group than CF3, with a pi-electron density of 0.972 at the para position versus 0.984 for CF3 (and 0.957 for the even stronger electron withdrawing group NO2) . See W.J. Hehre, L.Radom, and J.A. Pople, J. Am. Chem. Soc. 94:1496, 1972. https://pubs.acs.org/doi/abs/10.1021/ja00760a011
And I still do not understand why sigma acceptance of halogens outweigh pi donation making them deactivating group?
That is a simple question to ask, but not a simple question to answer.
Halogens are deactivating groups because the pi donation leads to a double bonded (and positively charged) Halogen. A positive charge on such electronegative atoms is so unstable that it outweighs the “stability” provided to the arenium ion by resonance of those pi electrons. Thus, the Halogen group is a weak deactivater.
No, the positive charge isn’t so bad, because it’s only a formal positive charge and not the positive charge resulting from less than a full octet. The reason halogens are deactivating is that they are highly electronegative, which withdraws electron density from the ring. Fluorine is much more deactivating than oxygen for this reason. When it comes to the heavier halogens, the problem is not only electronegativity but also poor orbital overlap – harder to form pi bonds between a 2p and a 3p orbital (for Cl) or 2p and a 4p orbital (for Br) and thus much weaker pi donors.
The oxygen in -OH group tend to donate its lone pair to form a pi bond, but the oxygen in -COR group tend to accept the pi electrons. Why?
In the latter case, the group directly attached to the aromatic ring is a carbonyl (C=O), which is a pi-acceptor. Draw it out and you will see.
Why is alkoxy put with amide and ester groups(in first category -nitrogens&oxygens with lone pairs-)???
If the atom directly attached to the aromatic ring is an oxygen, it will be an ortho-para director, regardless of whether that oxygen is part of an alcohol, ether, or ester functional group. The same applies to nitrogen re: amines and amides.
If the CARBONYL of an ester is directly attached to the aromatic ring, that’s a different story. In that case, it’s a meta director.
You are doing an amazing job.
Can’t thank you enough.
Truly amazing!
OK – thanks for stopping by, Saatvik!
sir, If we carry out the electrophilic aromatic chlorination of 2-methoxyphenol (say with NCS and PPh3S what will be the major product and why ?
The OH is the stronger donor (versus OCH3) so I would expect to see chlorination para to the OH.
One word: Amazing
Amazing website. Amazing tutorials. Amazing blog.
You make it all so simple and easy to understand.
Thanks a lot.
Can’t wait for the 2nd part. In the meantime, two small observations:
– text in 1st figure: “Deactivating groups tend to withdraw electron density to the ring” – shouldn’t it be “from the ring”?
– “bromination, chlorination, nitration, sulfonylation, Friedel-Crafts alkylation and Friedel-Crafts alkylation” – change one of the last two to “acylation”.