Enols and Enolates
Aldol Addition and Condensation Reactions
Last updated: February 16th, 2025 |
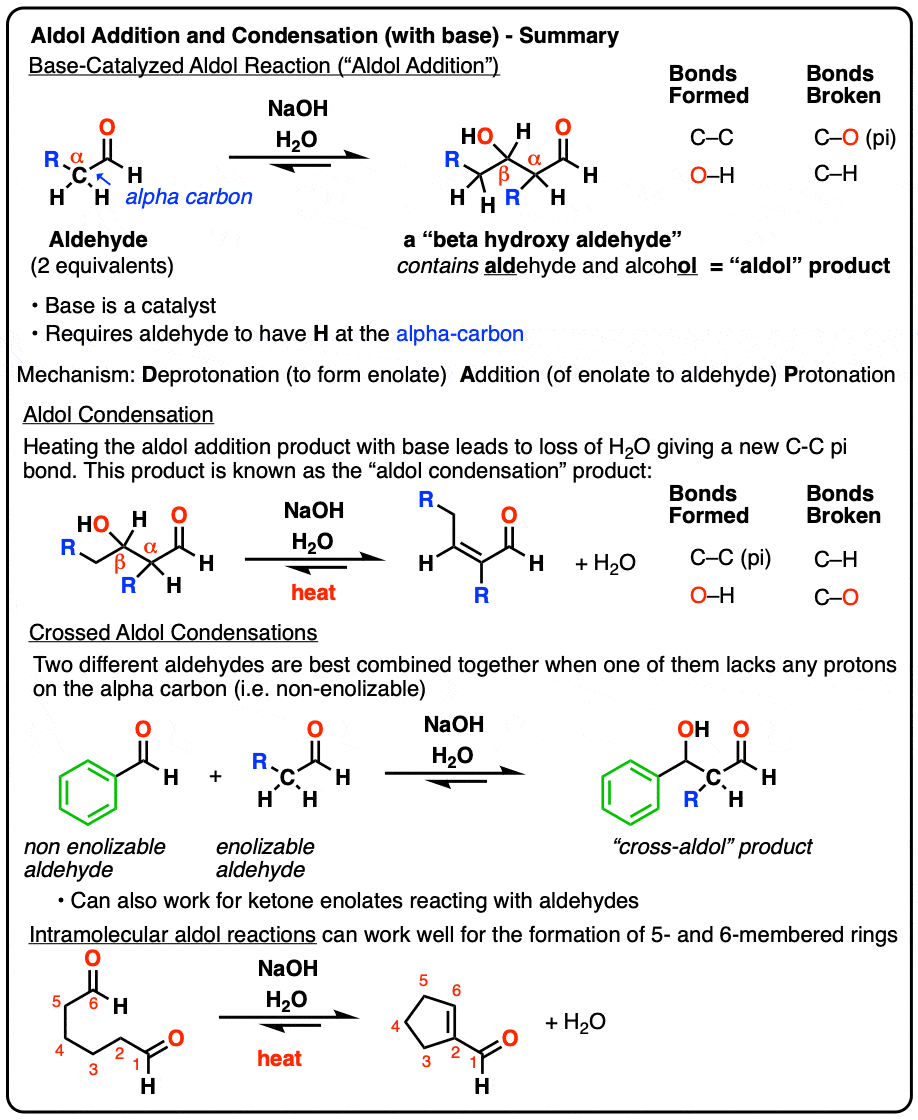
Aldol Addition and Condensation Reactions (Base-Catalyzed)
The Aldol Addition reaction is the addition of an enolate to an aldehyde (or ketone). Heating with base can result in loss of water to give a new C-C pi bond, giving a product we refer to as the Aldol Condensation product.
Similar reactions can proceed with the enolates of other species. They go by different names but the essential process is the same.
Table of Contents
- The Aldol Reaction (a.k.a. “Aldol Addition Reaction”)
- Mechanism of the Aldol Addition Reaction
- Aldol Condensation
- “Crossed” Aldol Reactions,
- Aldol Reactions of Ketones
- Intramolecular Aldol Reactions
- Aldol Reactions In Reverse – the “Retro Aldol”
- Summary
- Notes
- Bonus: More on the elimination step
- Bonus: Cousins of the Aldol Reaction (Knovenagel, Henry, Reformatsky)
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Aldol Addition Reaction
When certain aldehydes are treated with strong base, two molar equivalents of the starting aldehyde may form a new C–C bond. The resulting product, a “beta-hydroxy aldehyde” contains both an aldehyde and an alcohol.
In addition to the new C–C bond, a C-H bond has broken, as has a C–O (pi) bond:
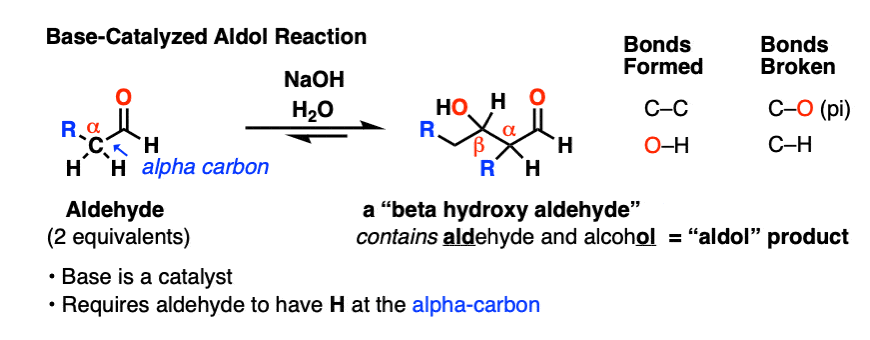
This reaction has come to be known as the Aldol reaction, or “aldol addition reaction” to distinguish them from cases where water is lost to give a new C-C double bond (that’s the aldol condensation, see part 3)
This type of reaction can also work for ketones (see below) and other functional groups as well (see appendix).
In this article we’ll focus on the base-catalyzed version of the aldol. An acid-catalyzed version also exists; it’s discussed [See post: The Acid-Catalyzed Aldol Reaction]
Here is a specific example of an aldol addition reaction, using 2 equivalents of ethanal (aka “acetaldehyde”).
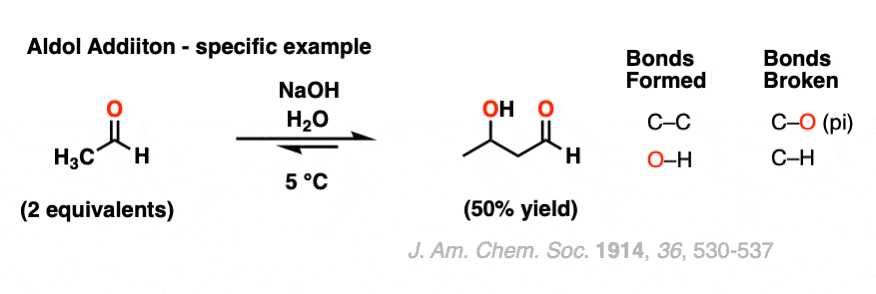
2. Mechanism of Aldol Addition Reaction
So how does this reaction work? What’s the purpose of the base? And how does that new C–C bond form?
When thinking about what a mechanism might look like, the first thing to note is that we are using a strong base (NaOH).
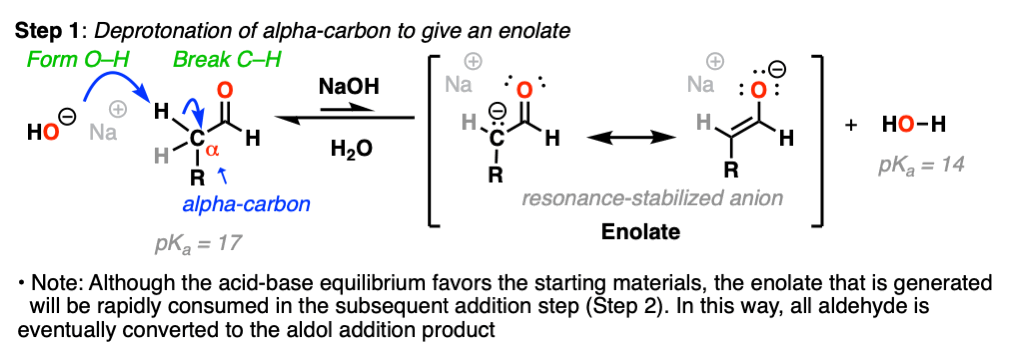
That begs a question: what’s the most acidic proton here? Hint: it’s not the C-H attached to the aldehyde! (that would result in an acyl anion, which is not resonance-stabilized).
Looking at the bonds that form and break, you should be able to see that we are breaking a C–H bond on the carbon directly adjacent to the aldehyde carbonyl (i.e. the “alpha carbon”).
Removal of this C–H bond by strong base results in a resonance-stabilized enolate ion. (Step 1, form O-H, break C-H).
Therefore, a necessary requirement for the aldol reaction is that the aldehyde be enolizable (i.e. have a proton on the alpha carbon). No proton → no enolate → no aldol.
hover to see some examples of enolizable vs non enolizable aldehydes or click on this link.
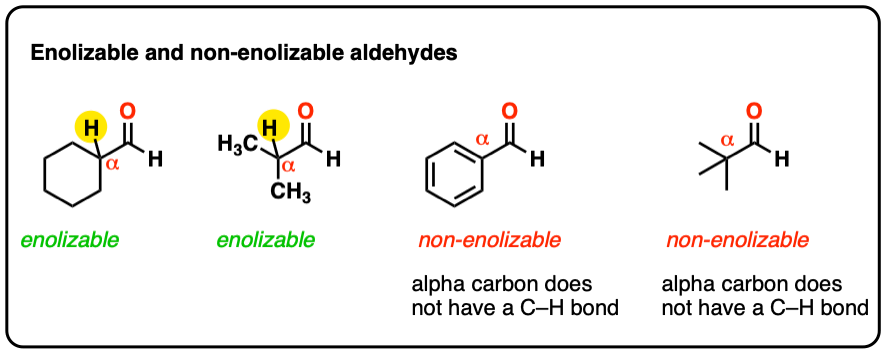{kind=link}
Note that acid-base equilibrium actually favors the starting materials – we are using NaOH (the conjugate base of water, pKa = 14) to make an even stronger base (the enolate, conjugate base of acetaldehyde, pKa = 17).
The key to the reaction is that the enolate, once formed, is an excellent nucleophile and can rapidly react with any available electrophiles hanging around in solution – such as, for example, aldehydes.
The product requires that we form C–C and break C–O (pi).
The enolate thus adds to the aldehyde in Step 2 (form C–C, break C–O (pi) ), an example of the most classic mechanistic step of carbonyls, addition.
In this way, all starting aldehyde is eventually converted to the aldol addition product, despite the unfavorable acid-base equilibrium.
After the addition reaction, the resulting negative charge on oxygen then undergoes protonation by the solvent (Step 3, form O–H).
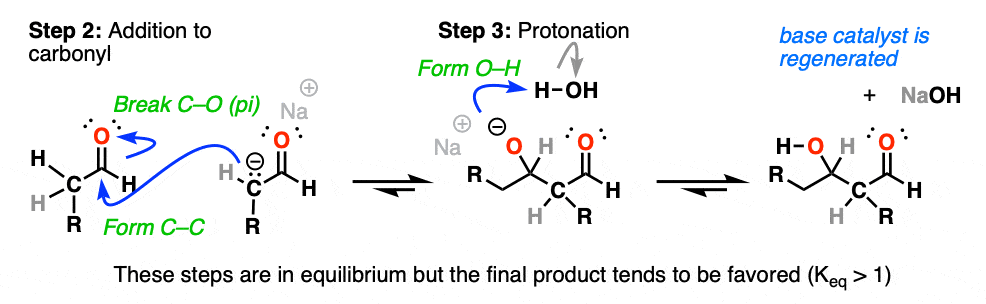
If you’ve previously covered the reactions of aldehydes and ketones, hopefully this sequence of events looks familiar.
It’s yet another example of the classic two-step mechanism (addition-protonation) to aldehydes, with the enolate as nucleophile.(OK, OK – 3 steps if you count enolate formation). [See: Aldehydes and Ketones – 14 Reactions That Follow The Same Mechanism]
Under these conditions, note that all 3 steps are in equilibrium. With aldehydes, equilibrium generally favors the final aldol addition product. [Note 1]
3. Aldol Condensation Reaction
But that’s not all! There’s more to the aldol than just a boring old addition reaction.
If the resulting aldol addition product (the beta-hydroxy aldehyde) is heated with base, it can lose water to give a new C-C pi bond.
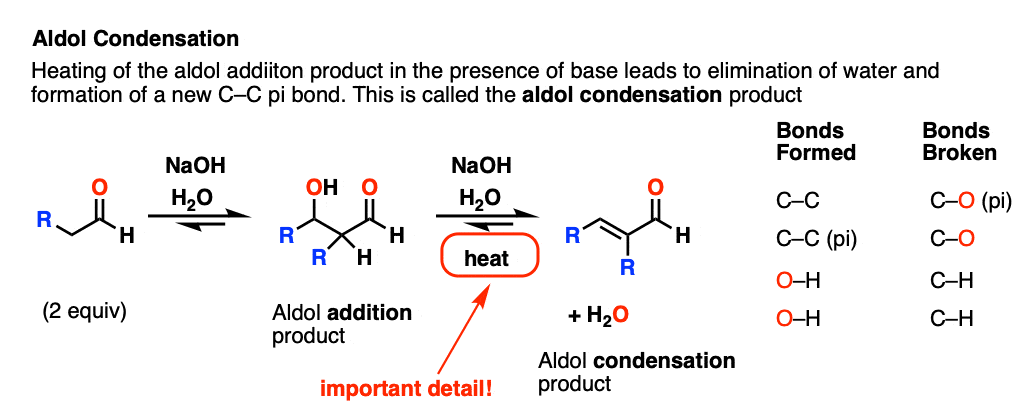
We’ve broken C-H, broken C-OH, and formed C-C (pi).
This is referred to as an aldol condensation reaction since water is formed as a byproduct.
For our purposes the most important distinguishing factor is “HEAT“, which tends to favor elimination reactions. [See: Elimination Reactions Are Favored By Heat]
A note for everyone reading this who expects to be tested on this material on an exam: if you see “heat”, assume that you will be expected to draw the condensation product.
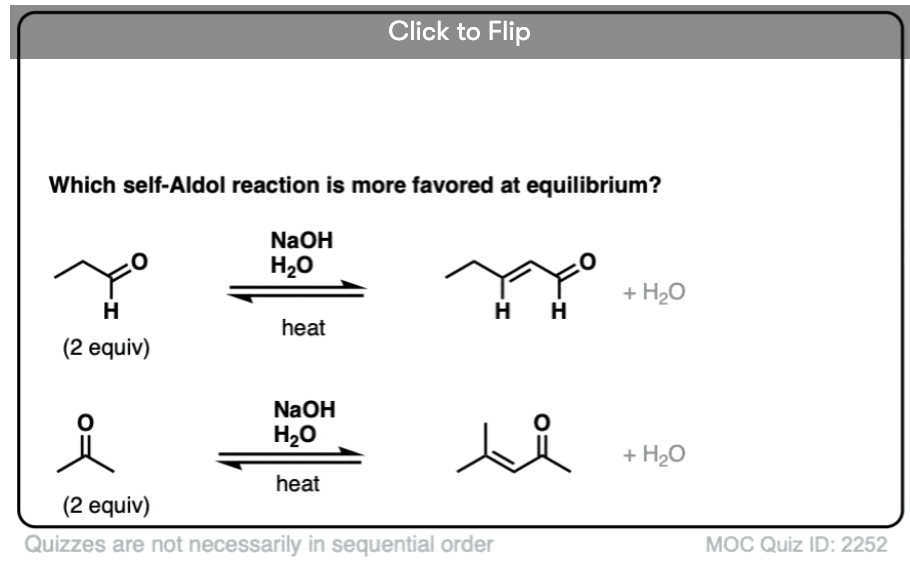
For two classic “real-life” examples, compare the major products for the same reaction run at low temperature (0-5 °C) versus “high temperature” (80 °C).
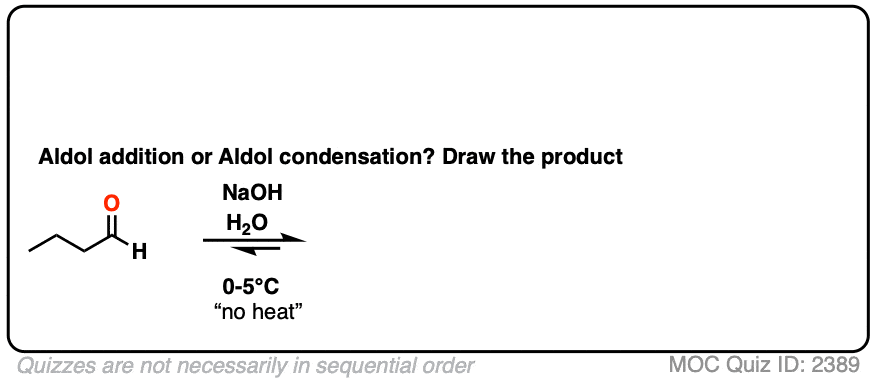 Click to Flip
Click to Flip
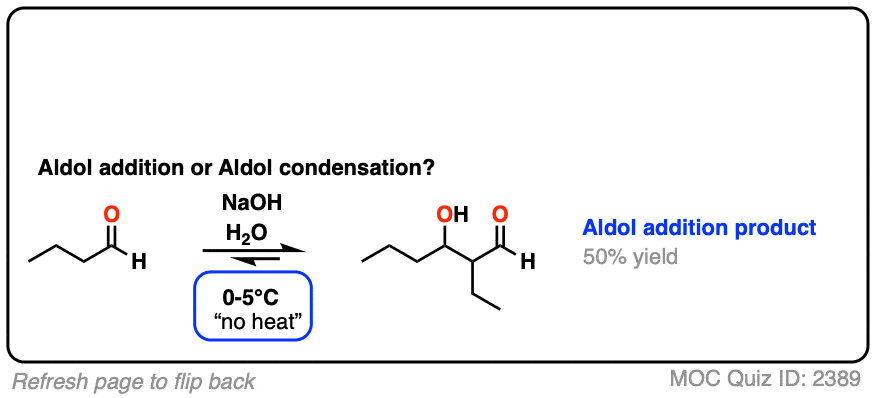
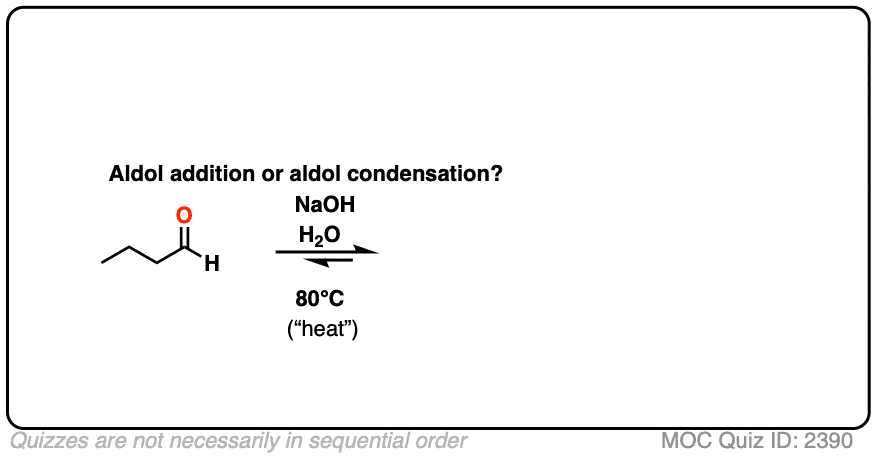 Click to Flip
Click to Flip
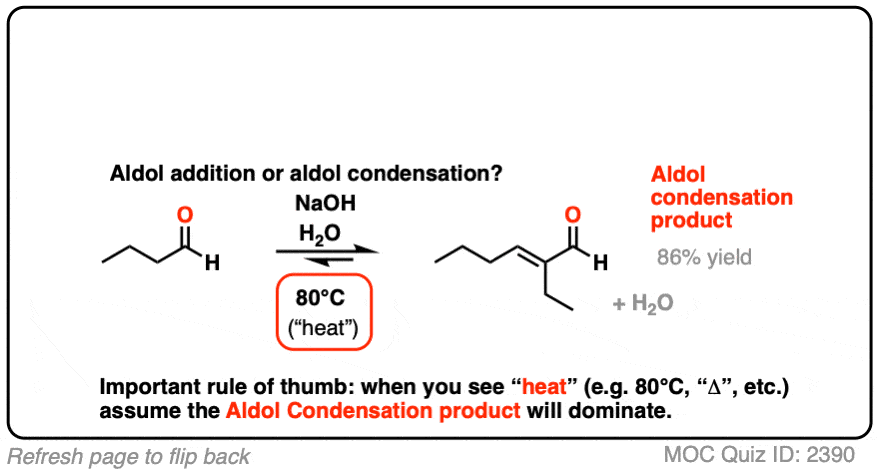
[See Note 2 for references]
In the real world, there are other factors which can determine the product distribution, but “heat” is the most important one from an “introduction to organic chemistry” perspective. [Note 3]
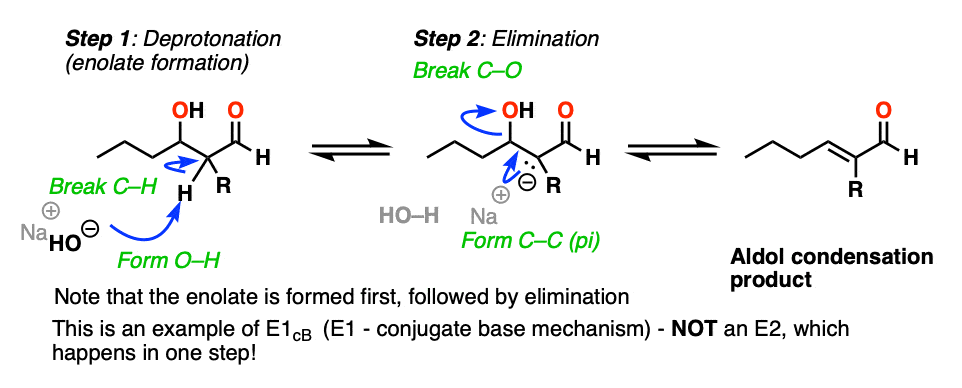
The elimination step works like this and is an example of an E1CB reaction – not an E2! For more discussion see the appendix [Note 4. ]
4. “Crossed” Aldol Reactions
So far, we’ve only dealt with “self-aldol” reactions, where two equivalents of the same aldehyde combine to give a new product.
So let’s take the next logical step. Why not combine together two different aldehydes?
For instance, let’s say I want to take the two aldehydes below and make the product on the right through an aldol reaction.
So I add the two aldehydes to solution, add my catalytic base, put up my feet, and wait for the reaction to complete, trusting in the Universe that it will all work out.
Nothing could possibly go wrong. Right?
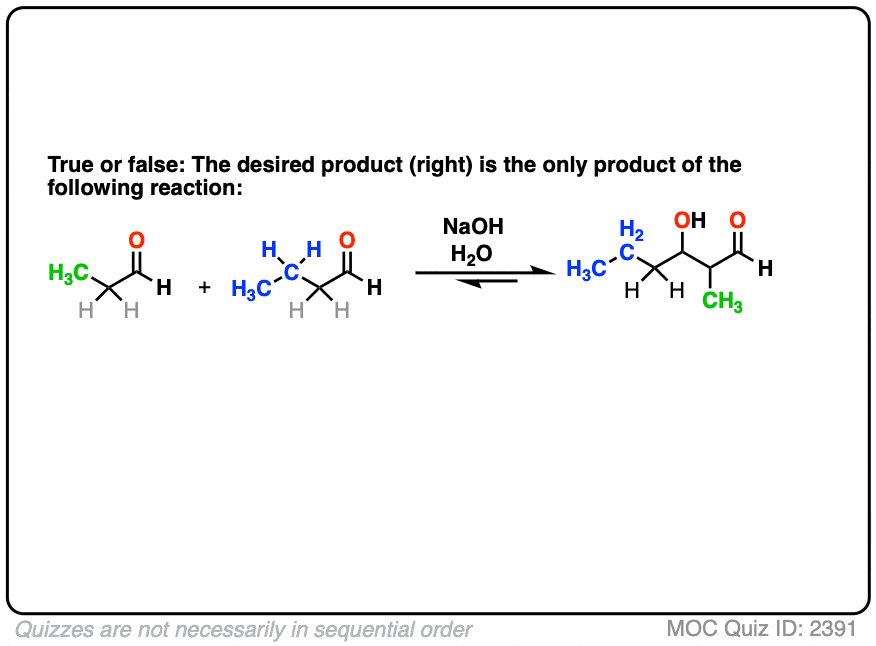 Click to Flip
Click to Flip
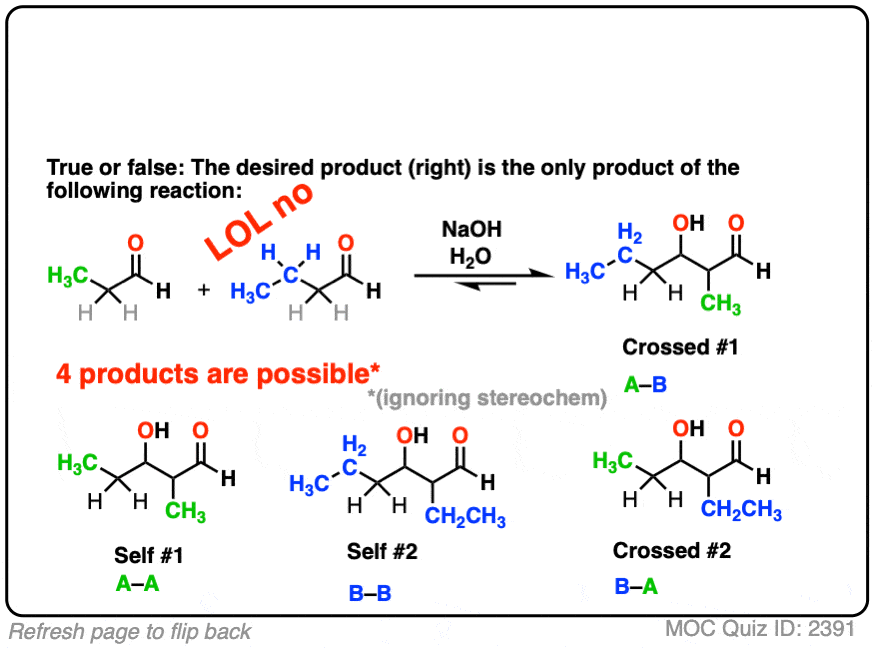
Just kidding of course. It’s chemistry. Lots of things could go wrong!
For one thing, if we have two enolizable aldehydes present in solution, then we can potentially form two different enolates.
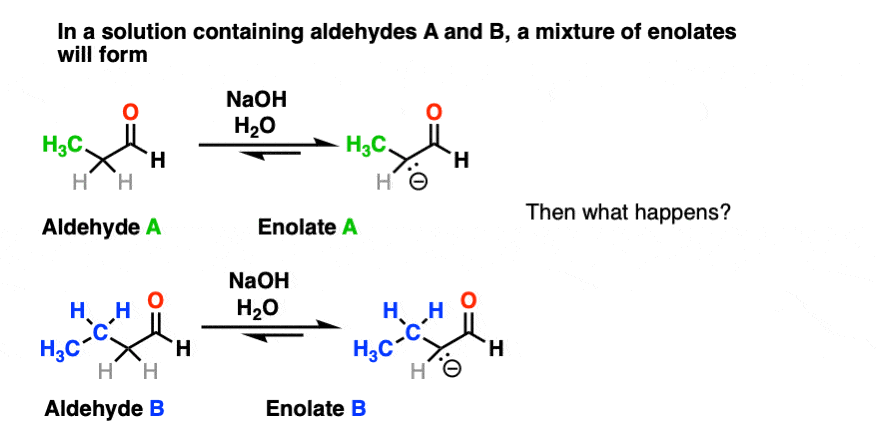
Secondly, since we are using catalytic base, there are plenty of aldehydes around.
Each enolate, in turn, may react with one of the two aldehydes, giving two “self-aldol” products and two “crossed aldol” products.
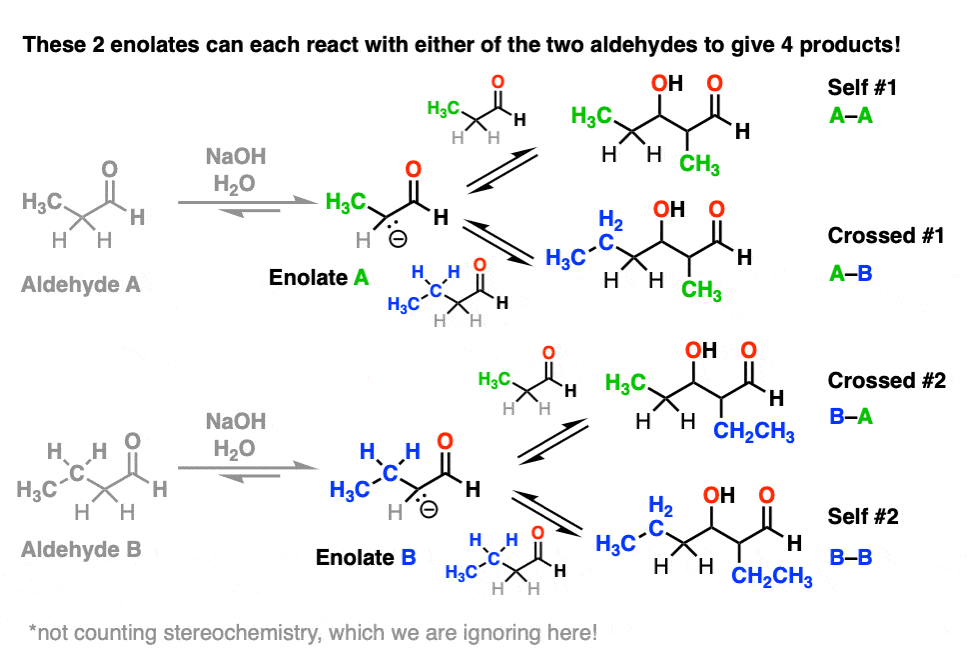
Not all of them will be formed in equal amounts, mind you, but this is still 4 products.
(And we’re making things easier by choosing to ignore stereochemistry here. [Note 5]
This result is what chemists in the business might call a BFM reaction. Euphemistically speaking, that stands for Big Frickin’ Mess. Although this real-life example (from 1914) actually turned out OK: [Note 6]
hover to see a real life example or click on this link.
{kind=link}
The fact is, nobody wants to run a reaction which produces 4 different products in significant yield. Separation of mixtures is a huge time-suck that every chemist tries to avoid as much as possible.
So how could something like this be done more efficiently?
One way is to employ a non-enolizable aldehyde as one of the reaction partners. That way there is only one enolate nucleophile to worry about.
This makes aldol reactions much more predictable – especially if we use an excess of the non-enolizable aldehyde to minimize self-reaction.
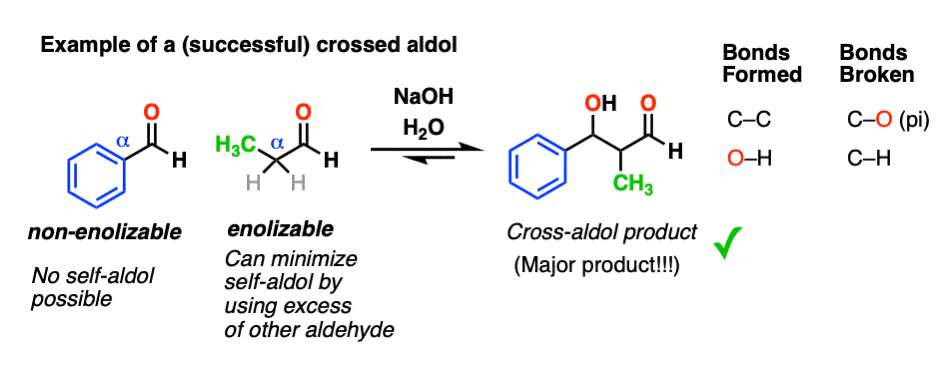
5. Aldol Reactions of Ketones
Enolates can also be formed from ketones, and ketones can also be the electrophiles in aldol reactions.
Here is the self-aldol reaction of acetone, for example.
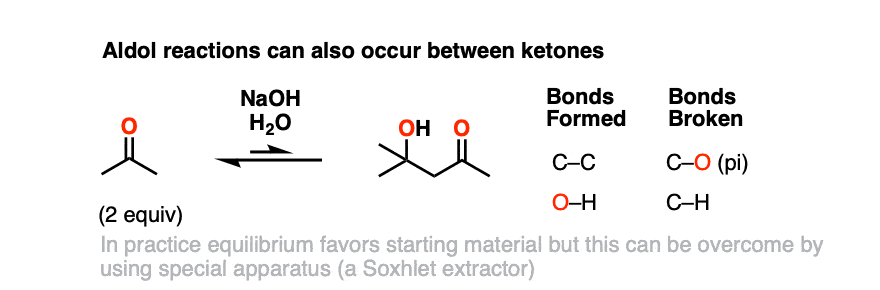
In practice, the self-aldol of acetone doesn’t work that well – equilibrium favors the starting materials – but using a device known as a Soxhlet extractor, it’s possible to isolate the product in reasonable yield.
While we’re on the topic it’s worth noting that ketones are generally less reactive than aldehydes towards nucleophiles, as the carbonyl carbon is more sterically hindered and also more electron rich (alkyl groups are electron-releasing – recall that they are “activators” in Friedel-Crafts reactions).
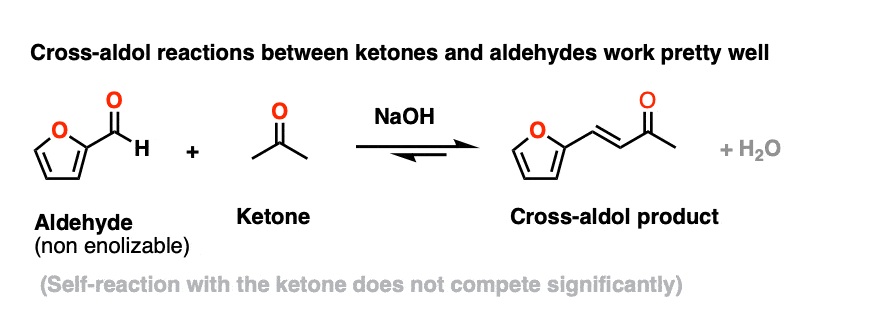
However, using a ketone enolate (formed with base) as a nucleophile for aldol reactions with non-enolizable aldehyde works extremely well. There are so many examples that this goes by its own name – the Claisen Schmidt condensation, not that you’d need to know that.
Here’s an example from Organic Syntheses. (Self-aldol with acetone does not compete to any significant extent)
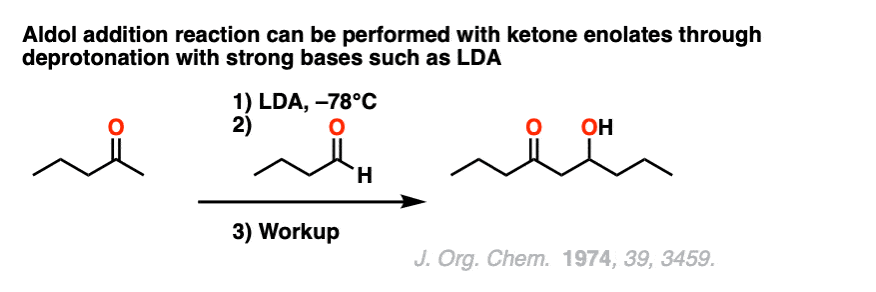
Another way to do aldol reactions with ketone enolates is to pre-form the enolate with a strong, bulkyl base such as lithium diisopropylamide (LDA) and then add the aldehyde.
[Note 7 ]
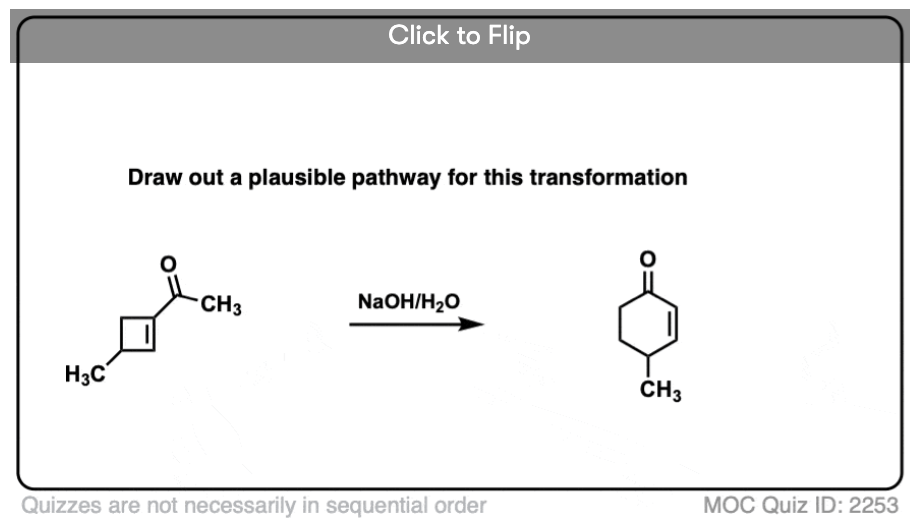
6. Intramolecular Aldol Reactions
I always tell students to expect to see intramolecular version of reactions on exams, since instructors know they are a great way of testing how well you understand a given reaction without them having to introduce any new concepts.
So I would be negligent if I omitted examples of intramolecular aldol reactions, which work best when a 5- or 6- membered ring can be formed. (Reminder – 5 and 6 membered rings have the least ring strain).
Look at this for instance:
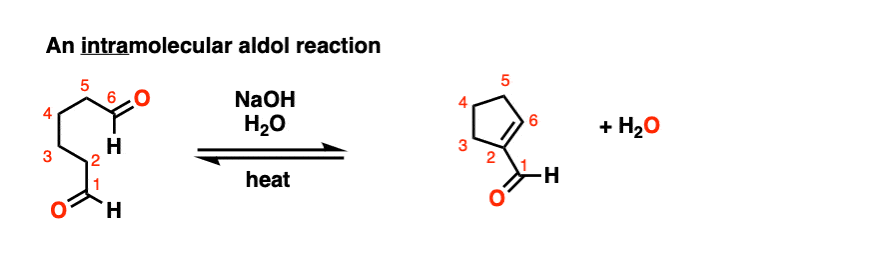
[Ref] Note there is no difference in the bonds that form or break from the intermolecular case (form C-C, break C-O (pi), break C-H). Since the newly-formed enolate and the aldehyde are on the same molecule, we will necessarily form a ring.
hover to see mechanism or click on this link.
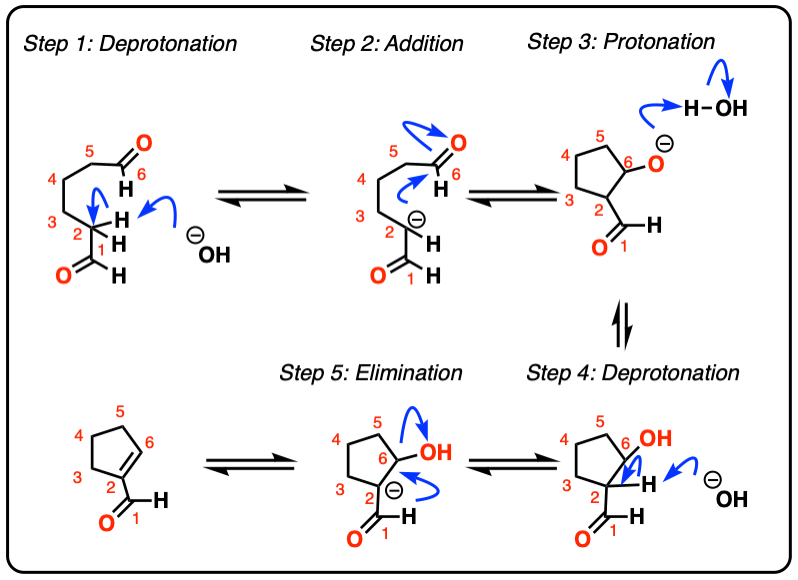{kind=link}
One common application of intramolecular aldol condensations is in the second step in the Robinson annulation reaction.
[Also: See Post: The Robinson Annulation ]
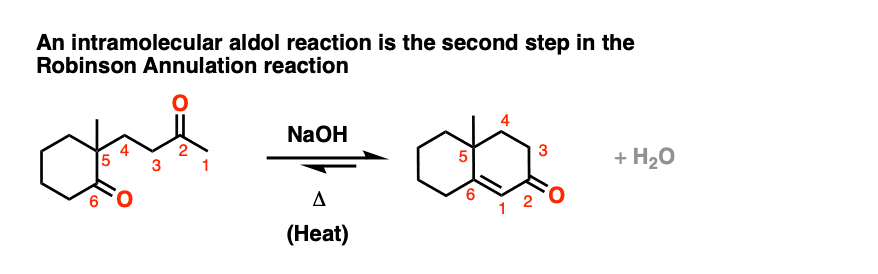
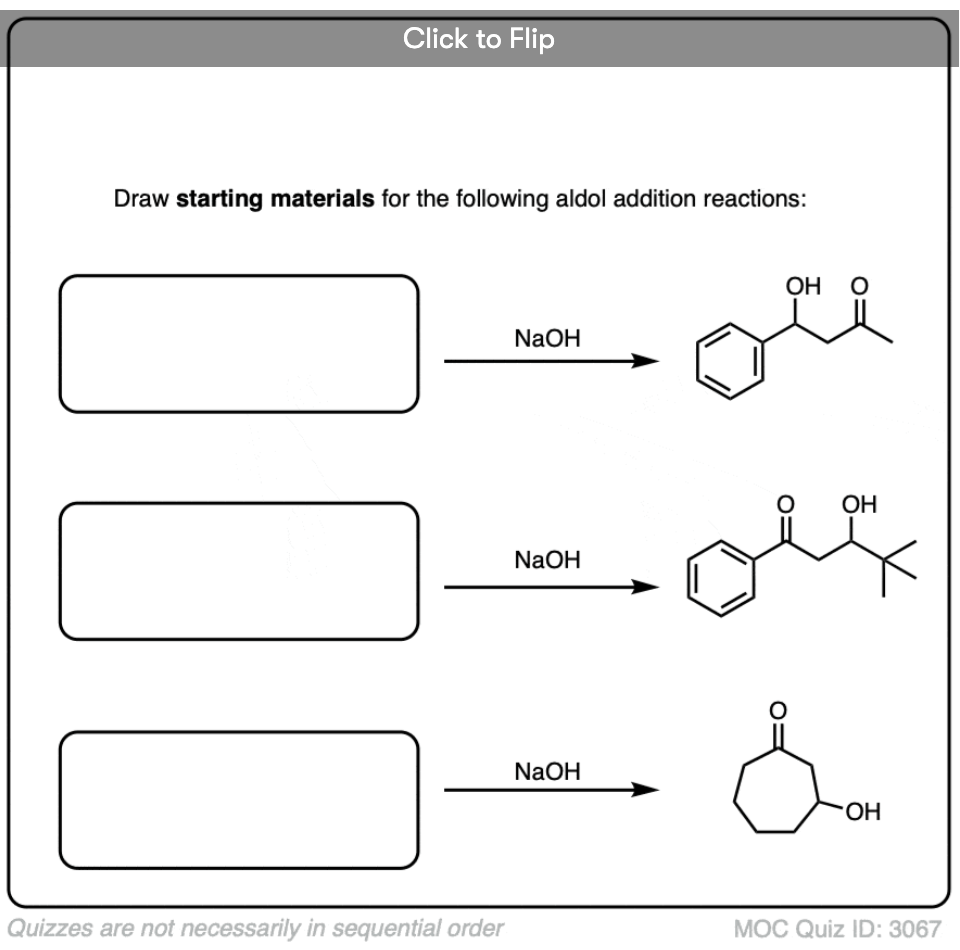
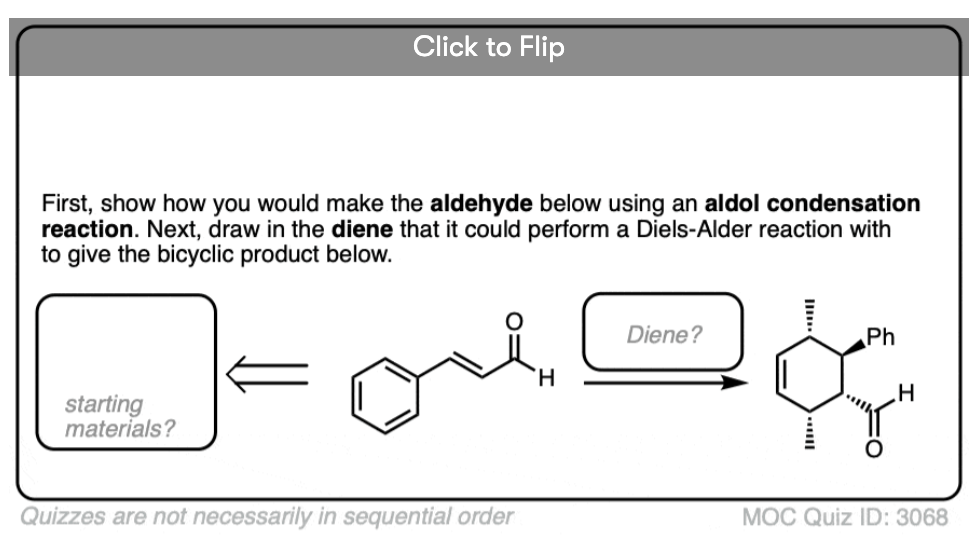
Several examples of intramolecular aldol reactions are in the “test yourself” section.
7. Retro-Aldol Reaction (Reversibility of the Aldol Reaction)
The aldol reaction is an equilibrium. Generally with aldehydes the reaction favors the final product as opposed to starting materials.
However sometimes it’s possible to get the reaction to work in reverse.
This is sometimes called the retro-Aldol reaction. Here is an example of a retro-aldol reaction driven by release of ring strain
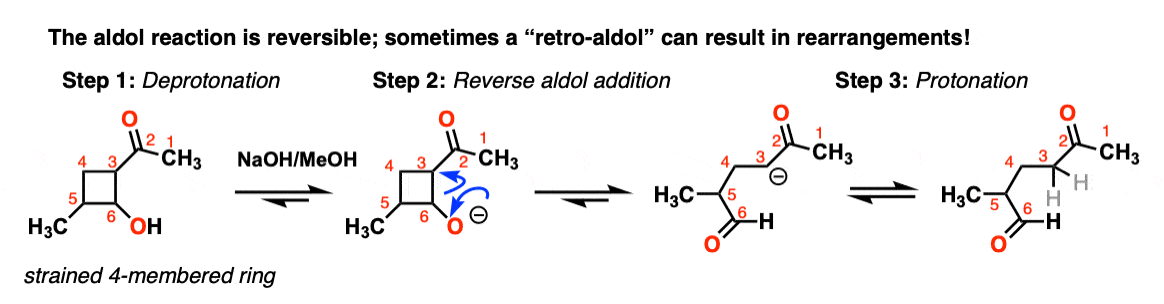
(note that the initial starting material was not made through an aldol).
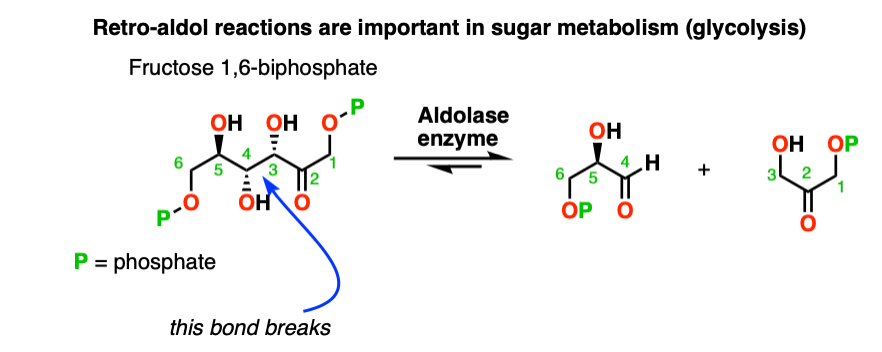
There are even enzymes (called “aldolases“) that can promote the retro-aldol reaction, such as in glycolysis. [Note 8]
8. Summary
- The base-catalyzed aldol addition reaction is the addition of an enolate to an aldehyde (usually).
- If heat is employed, expect a further loss of water and formation of a new double bond, leading to the aldol condensation product.
- Cross-aldol reactions work best when one enolizable aldehyde is used along with a non-enolizable aldehyde as the electrophile. Ketone enolates are also excellent nucleophiles for aldol reactions.
- The self-aldol reactions of ketones can occur, but they are much less favorable than those of aldehydes (the products are more sterically hindered).
- Watch out for intramolecular aldol condensation reactions, especially when 5- or 6- membered rings can be formed.
- “Retro-aldol” reactions are fragmentations of beta-hydroxy ketones or beta-hydroxy aldehydes that revert back to enolate / aldehyde starting materials.
- The aldol reaction can also be catalyzed by acid, where it proceeds through an enol intermediate. For more on that, see here.
Notes
Note 1. One study measured the equilibrium constant K to be about 400 [Ref]. The equilibrium constant decreases as steric hindrance in the product increases (e.g. as the aldehyde becomes more branched).
Note 2. The first reaction is from JACS 1914 36, 530. The second reaction is from Grignard & Vestermann, Bull Chem. Soc. Fr. Part 4, 1925, 37, 425. Hard to find online, but is referenced in Comprehensive Organic Synthesis.
Note 3. When an aromatic aldehyde is used it is difficult to get the reaction to stop at the addition stage, as the double bond will be considerably stabilized through conjugation with the aromatic ring.
Note 4. In a recent study of the mechanism of the aldol condensation, loss of the leaving group HO(-) was determined to be the rate-determining step. [Ref].
Note 5. We’re omitting stereochemistry in this post to keep things simple. I’ve also chosen to draw the enolate as a carbanion instead of the (more correct) planar geometry with a negative charge on oxygen.
With the enolate drawn this way, one particularly important detail is the geometry of the enolate, which can be either E or Z.
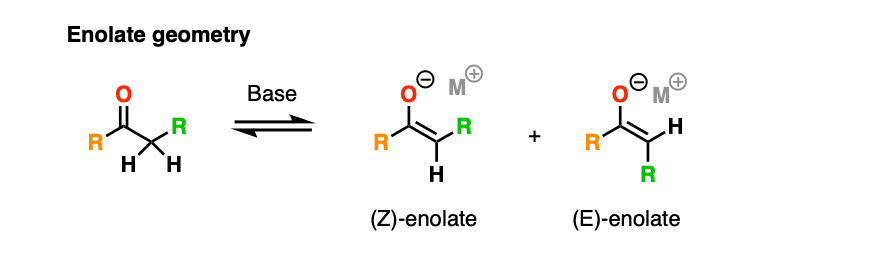
In 1957, Zimmerman and Traxler proposed a model for predicting the stereochemistry of aldol (and related reactions) through a closed, 6-membered chair-like transition state where the metal M coordinates to the oxygen of the enolate and the aldehyde.
In this model, the lowest-energy transition state tends to be the one where the bulky R groups occupy equatorial positions on the “chair”.
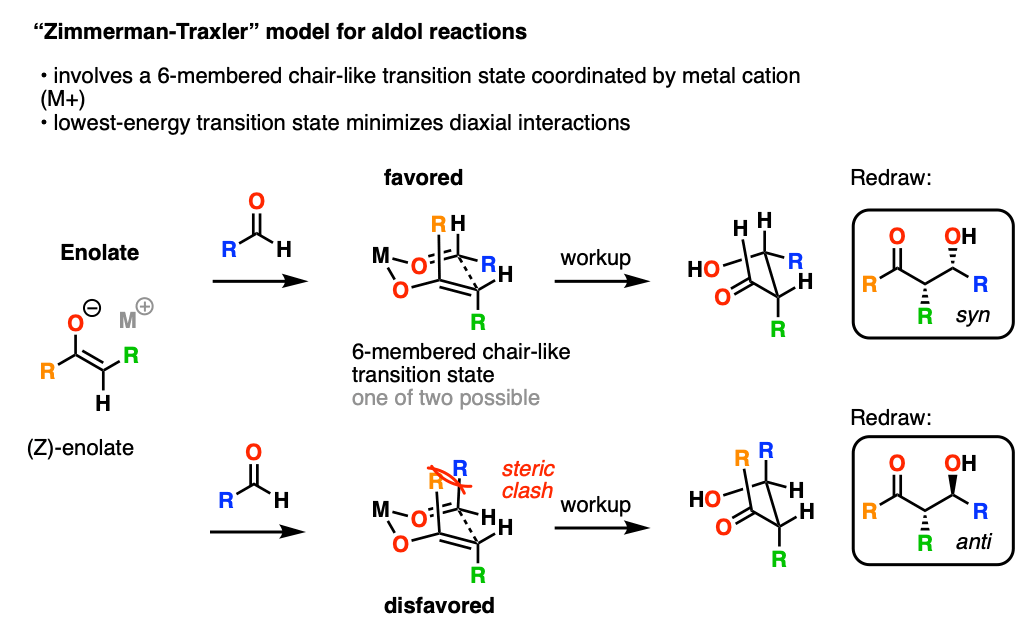
The metal M has a huge effect on stereoselectivity. The shorter the O-M bonds, the more compressed the “chair” transition state will be, and the greater will be the stereoselectivity. In particular, aldol reactions with boron enolates have shown spectacular stereoselectivity.
Note 6. This example is from 1914, before modern spectroscopic and separation techniques made identifying all the reaction products feasible. The major product was distilled in 54% yield, which is actually a pretty good result! An old generalization called the “Lieben rule” stated that the major aldol product results from attachment of the “more substituted” enolate to the “less substituted” aldehyde.
Note 7. Here is a classic example of an aldol reaction between a ketone enolate and an aldehyde using lithium di-isopropylamide (LDA) to form the “less substituted” enolate. [Ref]
Note 8. Retro aldol reactions are actually part of sugar metabolism! In the body, 1,6-fructose biphosphate is cleaved to through the action of an aldolase, giving glyceraldehyde 3-phosphate and dihyroxyacetone phosphate.
The Elimination Step In The Aldol Condensation Reaction
So how does the”elimination” step work?
First of all, why is HO(-) a leaving group? Isn’t it a strong base, and therefore a bad leaving group? Yes, it is a strong base, and generally not a good leaving group in elimination reactions. However, this is not a typical elimination reaction.
The first instinct might be to think of this as an E2 reaction, since we’re using strong base.
This is actually not an E2 reaction because it’s not concerted!
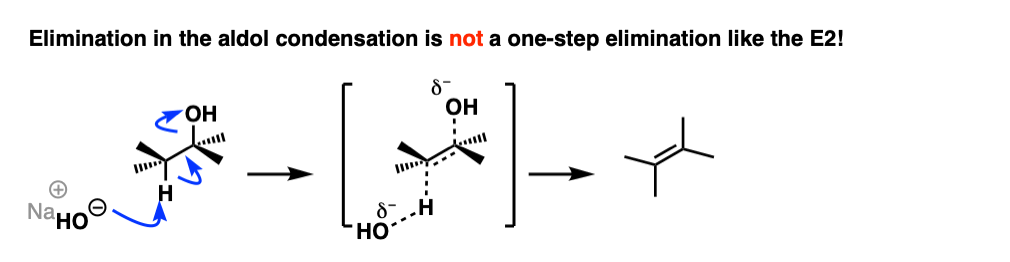
Instead this is an example of E1CB (elimination, unimolecular rate determining step, via the conjugate base). [See post: The E1cB mechanism]
In the presence of strong base (e.g. hydroxide, conjugate base of water, pKa 14), an enolate can be formed from the beta-hydroxy aldehyde (pKa of around 17-18) .
Note that the enolate product is actually a stronger base than the hydroxide ion, so equilibrium lies on the side of the starting materials.
The enolate is an intermediate that can be observed and isolated (unlike the bimolecular transition state in the E2!).
Now, if you think of the negative charge as being stuck on the carbon on a p-orbital, imagine that p-orbital overlapping with the adjacent C-O sigma* bond. If the overlap is sufficient, then a new C-C (pi) bond can form along with breakage of the C-OH bond.
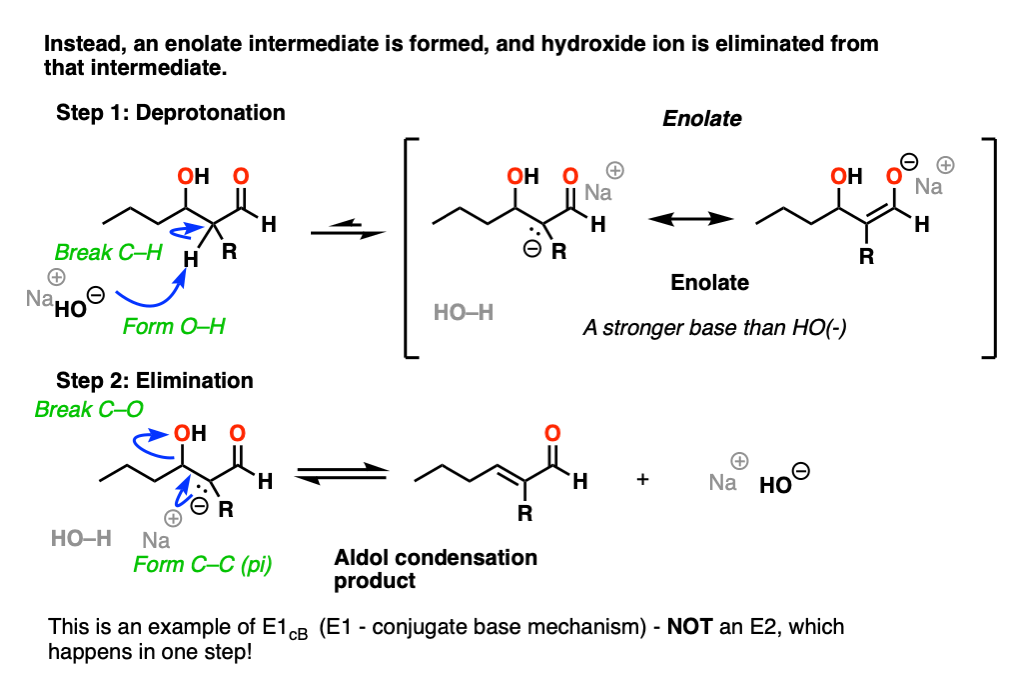
So in this second step, we’re actually going from a stronger base (the enolate) to a weaker base (hydroxide).
So yes, hydroxide ion is a strong base and a bad leaving group, but the reaction to expel NaOH is actually favored from an acid-base perspective (where stronger acid + stronger base –> weaker acid + weaker base).
Furthermore, the product has a new double bond in conjugation with the C=O bond, which is stabilizing.
Cousins of the Aldol Reaction – Knovenagel, Henry, Reformatzky, and Other Variants
In this article we’ve discussed the reaction of enolates of aldehydes and ketones, but it’s possible for other “enolates” to form too. This should probably be a separate article, but the mechanisms are almost all very similar to those of the aldol so they are treated briefly here. (Step 1 – formation of enolate; Step 2 – addition to carbonyl, then Step 3 – protonation, followed by elimination in some cases).
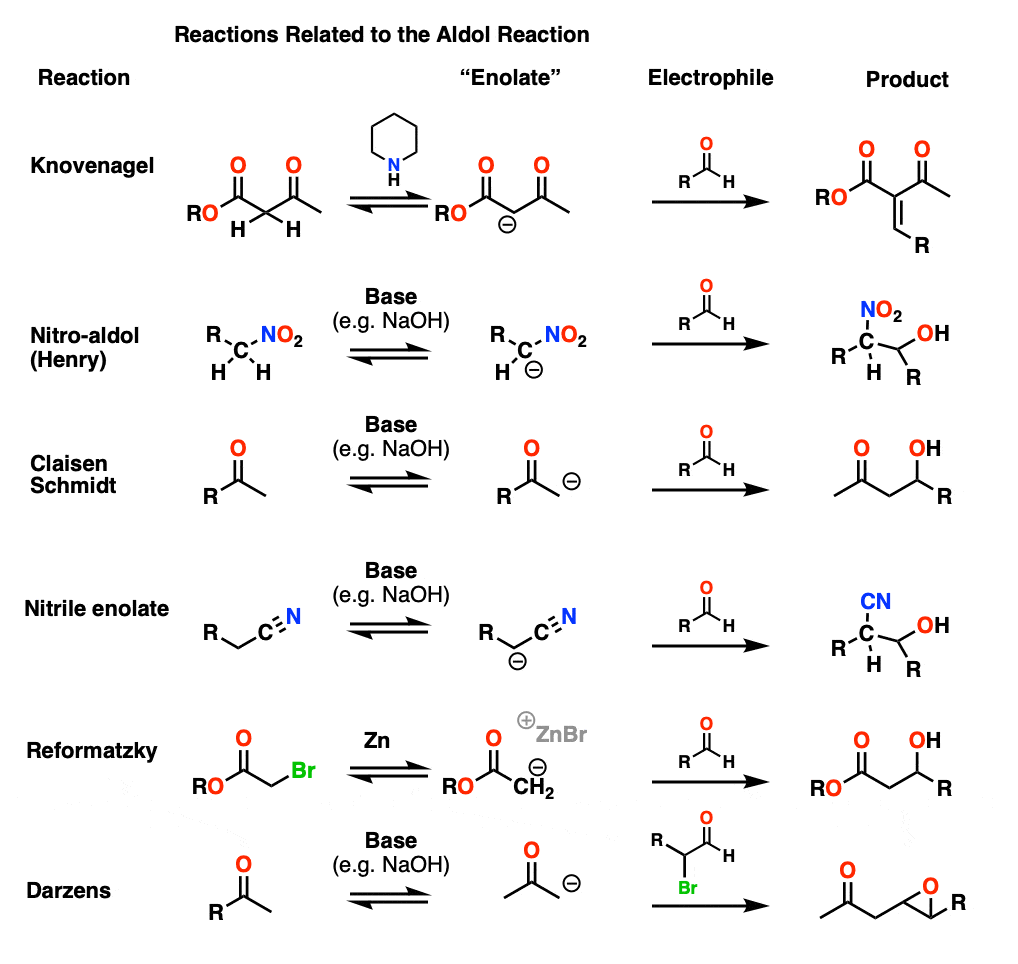
Knovenagel condensation
In the Knovenagel condensation, a beta-keto ester (or similar) compound is treated with a mild base (piperidine is often used) to form the enolate, which can then attack an aldehyde. Loss of water is usually very rapid since the alpha proton is generally quite acidic (pH around 12).
Henry reaction
In the nitro-aldol or Henry reaction, the carbon adjacent to a NO2 group is deprotonated to give a species much like an enolate.
Claisen-Schmidt
In the Claisen-Schmidt (name is not important to remember) a ketone enolate is formed and adds to an aldehyde.
Nitrile enolates
The carbon adjacent to nitriles may also be deprotonated although with pKa about 30, using NaOH is a stretch. In essence this is no different than a typical aldol reaction.
Reformatsky Reaction
In the Reformatsky, one starts with an alpha-bromo ester and then treats it with finely divided zinc dust. This is very much like forming a Grignard reagent, except that the resulting carbanion is an enolate! The enolate can then add to an aldehyde or ketone.
Darzens Reaction
This is a slightly weird one, an aldol with an alpha-halo aldehyde as electrophile. After addition of the enolate, the resulting alkoxide can then form an epoxide via SN2 displacement of the C-Br bond (See this post on Synthesis of Epoxides). Basically, the aldol reaction makes a haloether, which then snaps shut to give an epoxide.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
(Advanced) References and Further Reading
The review by Heathcock in Comprehensive Organic Synthesis, vol. 2. pp 139-177 was extremely useful in finding useful literature references on the aldol reaction. Recommend.
- Ueber die Einwirkung des Natriums auf Valeraldehyd
A. Borodin
J. Prakt. Chim. 1864 93 (1), 413-425
DOI: 10.1002/prac.18640930168
The first publication in the field of ‘aldol’ chemistry, by Aleksandr Borodin, all the way back in 1864. This article involves treatment of valeraldehyde (C5H10O) with sodium to give what we would now call aldol condensation products. A subsequent report in 1872 involved the treatment of butanal with HCl to obtain products similar to those reported by Wurtz.
This article has historical details on Borodin’s colorful life; besides being known for chemistry, he was also a popular composer and his works are still played today. - Sur an aldéhyde-alcool
Wurtz, M. A.
Bull Chim. Soc. Fr. 1872, 436
No DOI, but here is full LINK
Adolphe Wurtz examined the treatment of ethanal with HCl at low temperature and obtained a new “aldehyde-alcohol” product C4H8O2 . “Ce corps est l’aldehyde-alcool qui fair l’objet de cette note et que je nomerai par abreviation aldol.” The name “aldol” has been with us ever since. - Condensationen von Ketonen mit Aldehyden [Condensations of ketones with aldehydes]
Claisen, L.; Claparède, A.
Ber. 1881, 14 (1): 2460–2468
DOI: 10.1002/cber.188101402192 - “Ueber die Einwirkung von Aceton auf Furfurol und auf Bittermandelöl in Gegenwart von Alkalilauge” [On the effect of acetone on furfural and on bitter almond oil (benzaldehyde) in the presence of alkali hydroxides]
Schmidt, J. G.
Chem. Ber. 1881, 14 (1): 1459–1461
DOI: 10.1002/cber.188101401306
The reaction between an aldehyde or ketone having an α-hydrogen with an aromatic carbonyl compound lacking an α-hydrogen is called the Claisen–Schmidt condensation. This reaction is named after two of its pioneering investigators Rainer Ludwig Claisen and J. G. Schmidt, who independently published on this topic in 1880 and 1881. - The Complete Mechanism of an Aldol Condensation
Charles L. Perrin and Kuei-Lin Chang
The Journal of Organic Chemistry 2016, 81 (13), 5631-5635
DOI: 10.1021/acs.joc.6b00959
A very nice mechanistic study of the base-catalyzed crossed aldol reaction between acetophenone and benzaldehyde, forming chalcone. This paper establishes that the final elimination of HO(-) is the rate-limiting step in this particular reaction through observing that the reaction is faster in D2O than H2O (a solvent isotope effect). - Studies on compounds related to auxin-a and auxin-b. Part III. The preparation and properties of the cyclopentenyl analogue of auxin-b lactone
J. B. Brown,, H. B. Henbest, and E. R. H. Jones.
J. Chem. Soc. 1950, 3634-3641
DOI: 10.1039/JR9500003634
Nice example of an intramolecular aldol condensation here. - “Mixed condensations of two ketones are rarely preparatively useful.” from Heathcock, C. H. in Comprehensive Organic Synthesis vol. 2. section 1.5 “The Aldol Reaction: Acid and General Base Catalysis”. pp 133-179. See Guthrie’s paper, below, where the equilibrium constant for aldol condensation is 0.039 (i.e. favors starting materials). It is possible to get decent yields by sequestering the aldol product from the base with a Soxhlet apparatus.
- The Aldol Condensation of Acetaldehyde: the Equilibrium Constant for the Reaction and the Rate Constant for the Hydroxide Catalyzed Retro-Aldol Reaction
J. Peter Guthrie
Can. J. Chem. 52, 2037 (1974). f
DOI: https://doi.org/10.1139/v74-294
“An indirect thermochemical estimate of the equilibrium constant for the aldol condensation of acetaldehyde suggested that this reaction was much less irreversible than has been believed… the equilibrium constant is 4.0 × 102 M-1…. the enthalpy change is –9.84 kcal/mol.” [A calculation for ΔG° based on these parameters gives -3.55 kcal/mol]
A similar study on the aldol condensation of acetone leads to an equilibrium constant of 0.039 (i.e. favors the starting materials) and a value for ΔG° of + 1.92 kcal/mol. Ref. - The Stereochemistry of the Ivanov and Reformatsky Reactions. I
Howard E. Zimmerman and Marjorie D. Traxler
Journal of the American Chemical Society 1957 79 (8), 1920-1923
DOI: 10.1021/ja01565a041
The Zimmerman-Traxler transition state model for aldol reactions, which has proved extremely useful in rationalizing (and predicting) stereochemical outcomes. - The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes
Alan B. Northrup and David W. C. MacMillan
Journal of the American Chemical Society 2002 124 (24), 6798-6799
DOI: 10.1021/ja0262378
A cross-aldol reaction of two aldehydes that goes through an enamine rather than enolate intermediate, using the amino acid L-proline as a catalyst.
Real-Life Examples:
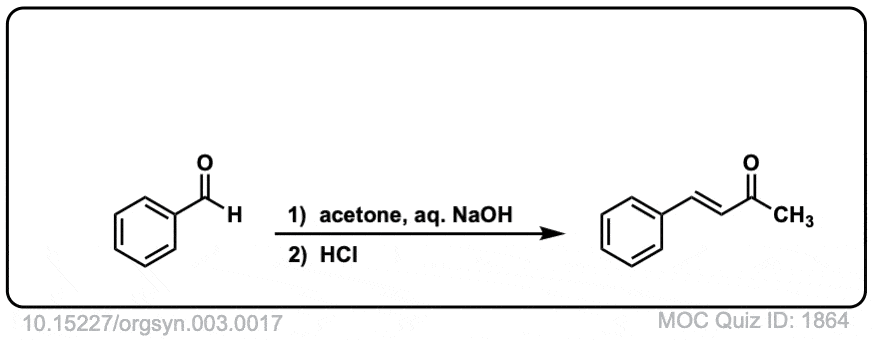
Org. Synth. 1923, 3, 17
DOI Link: 10.15227/orgsyn.003.0017
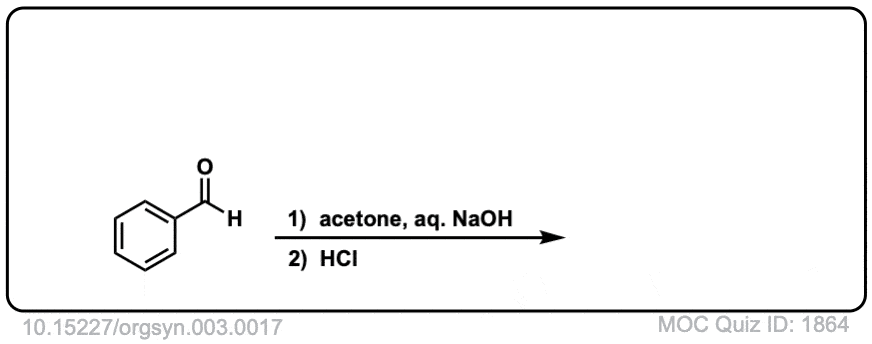 Click to Flip
Click to Flip
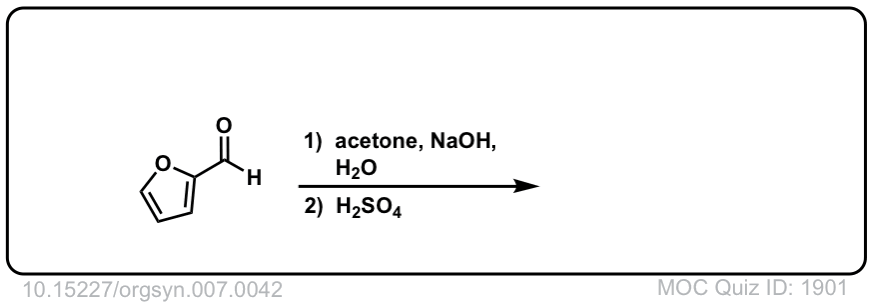
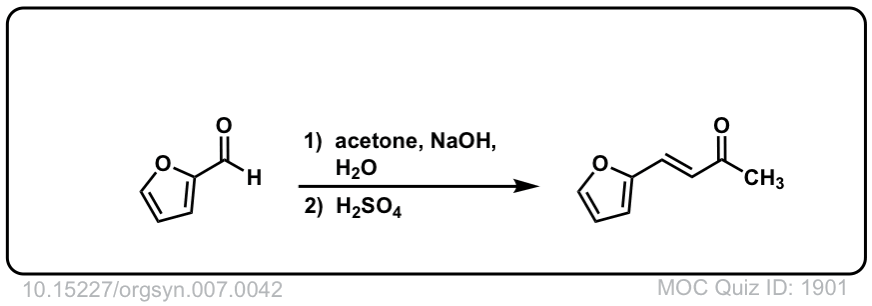
Org. Synth. 1927, 7, 42
DOI Link: 10.15227/orgsyn.007.0042
 Click to Flip
Click to Flip
Org. Synth. 1932, 12, 22
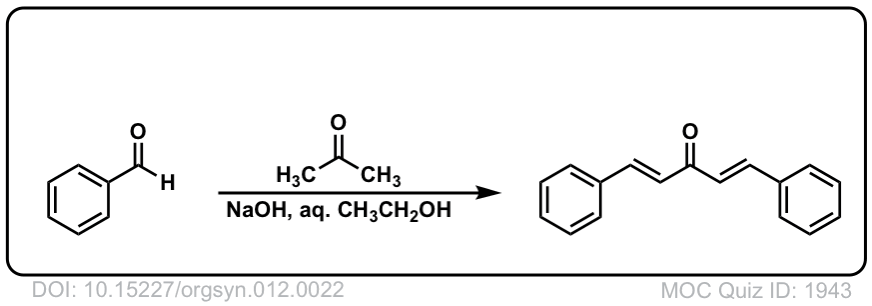
DOI Link: 10.15227/orgsyn.012.0022
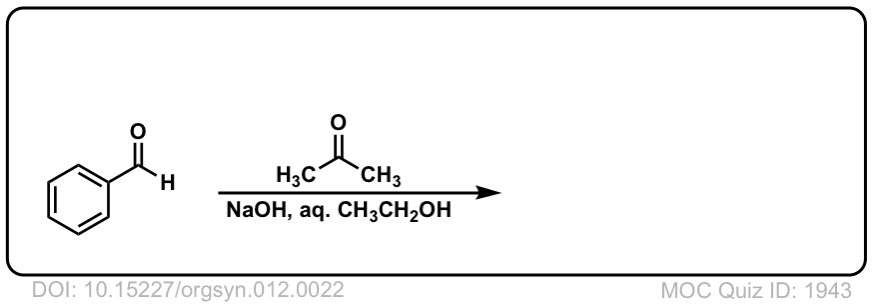 Click to Flip
Click to Flip
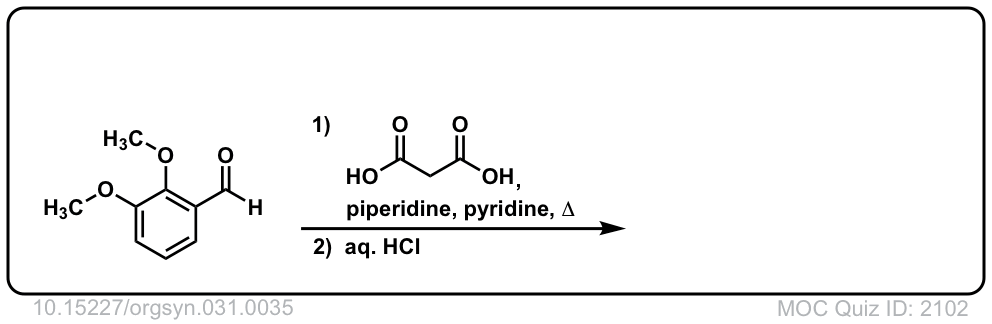
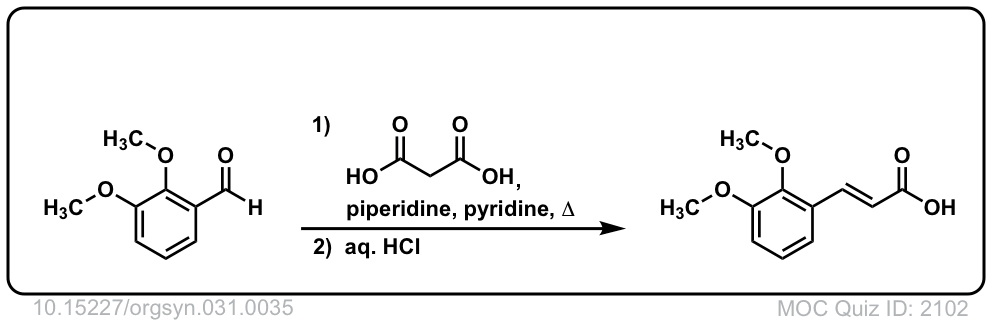
Org. Synth. 1951, 31, 35
DOI Link: 10.15227/orgsyn.031.0035
 Click to Flip
Click to Flip
Hello, I’d like to make a remark. The statement “So in this second step, we’re actually going from a stronger base (the enolate) to a weaker base (hydroxide).
So yes, hydroxide ion is a strong base and a bad leaving group, but the reaction to expel NaOH is actually favored from an acid-base perspective (where stronger acid + stronger base –> weaker acid + weaker base).” is not true. It is a bit more complex than this. If this was true, the Wolff–Kishner reduction wouldn’t take place and SN2 reactions of amines and ethers with BuLi would take place. Thanks for all the info on the aldol reaction, anyway.
I didn’t say that acid-base equilibria always dictate the outcome of reactions. I just said that it’s a factor in this case.
A common question I get on this step is “wait, I thought HO(-) is a strong base and a poor leaving group” and to address this potential objection, my rationalization is that HO(-) is not a strong base *relative* to the enolate.
Of course reactions are more complex than that. I would absolutely not take it as far as to say that SN2 reactions of amines and ethers with BuLi would take place.
Thanks for Note 8 about aldolase. I was confused about how 1,6-fructose bisphosphate was an aldol because textbooks always draw it in ring form. Makes a lot of sense, now!
this why i use this site
Thank you. Glad it cleared things up!
If we got a methyl ethyl ketone, and have to perform aldol (by treating with OH-) , I think we will form form the thermodynamic enolate and then proceed. But my instructor states that the methyl’s carbon will get deprotonated because then we will get a 1° degree carbanion that will be more stable than 2°.I am super confused, please help me out.
Hello, below the notes (The Elimination Step In The Aldol Condensation Reaction) you made the arrow toward the enolate longer than the arrow in the opposite direction. it’s a mistake right ?
that LOL NO was epic lmao. ill never forget cross aldol again
So powerful and informative!
Glad you find it helpful Robert!
Thanks for Note 8 about aldolase. I was confused about how 1,6-fructose bisphosphate was an aldol because textbooks always draw it in ring form. Makes a lot of sense, now!
Glad you found it helpful Alexander – 99% of people don’t read down to the notes, but I am thankful for the small percentage (like you) who do!
Very amazing 🤩👍👍