Enols and Enolates
The Michael Addition Reaction and Conjugate Addition
Last updated: February 6th, 2025 |
The Michael Addition Reaction (and Conjugate Addition)
- The Michael reaction (sometimes “Michael Addition”) is the addition of an enolate to an electrophilic alkene, such as an alpha-beta unsaturated ketone, nitrile, or ester
- The primary driving force is formation of a C-C (single) bond (80 kcal/mol) and breakage of a C-C pi bond (60 kcal/mol)
- The mechanism for the Michael reaction is 1) deprotonation to form an enolate, 2) conjugate addition of the enolate to the alkene, and 3) protonation of the resulting (new) enolate
- Conjugate addition (or “1,4-addition”) is the generic name for reactions where a nucleophile adds to the beta-carbon of electrophilic alkenes that are in conjugation with an electron-withdrawing group
- Nucleophiles that can participate in conjugate addition include enolates, amines, thiolates [RS(-)], enamines and dialkylcuprates (“Gilman reagents”)
- Grignard reagents do not perform conjugate addition; they add to carbonyl carbons instead (“1,2-addition”), as do organolithium reagents and LiAlH4.
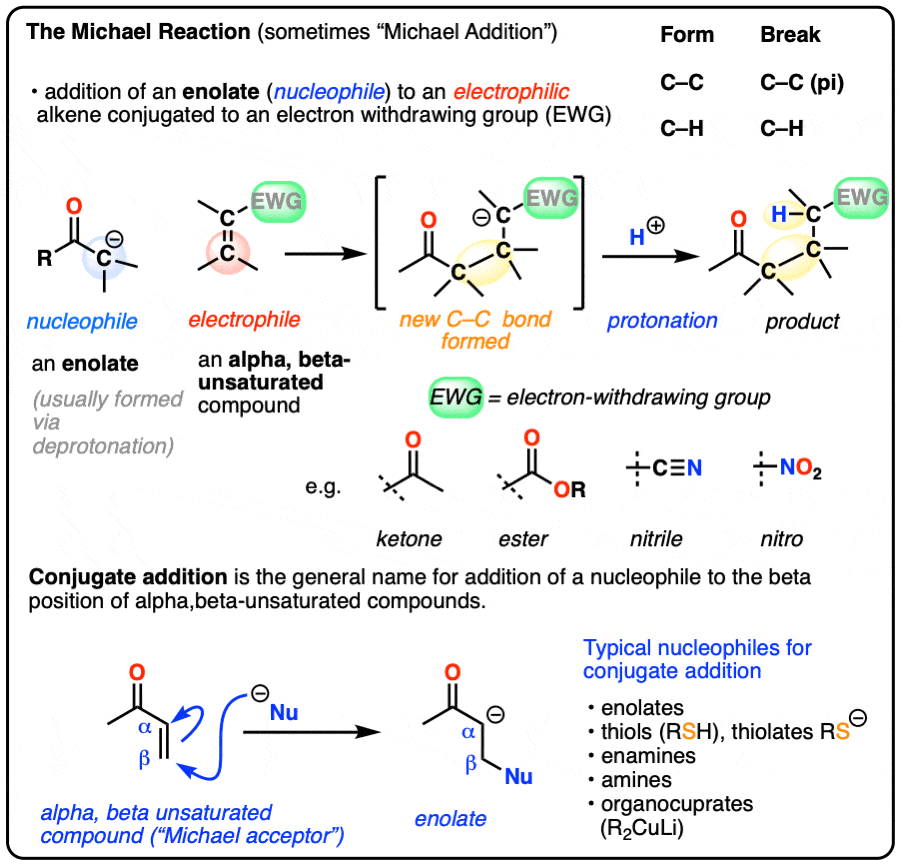
Table of Contents
- The Michael Reaction – Some Examples
- General Pattern of the Michael Reaction
- Mechanism of the Michael Reaction
- Why Don’t Nucleophiles Add To “Normal” Alkenes?
- Conjugate Addition (“1,4-Addition”)
- 1,2-Addition vs. 1,4-Addition
- Intramolecular Michael Reactions
- Working Backwards From Michael Addition Products
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. The Michael Reaction – A Few Examples
Enolates can add to the beta (β) carbon of alpha-beta carbonyl compounds, forming a new C-C bond and breaking a C-C pi bond. This is known as the “Michael Reaction” in honor of Arthur Michael’s initial (1887) report of this reaction [Ref]. (See article – Enolates – Formation, Stability and Reactions)
Since a C-C pi bond is being broken and two new single bonds are being formed to those carbons, the Michael reaction is an example of an addition reaction. (See article – Addition Reactions – Elimination’s Opposite)
Here is an example of a Michael reaction between the enolate of a malonic ester and an alpha, beta-unsaturated ketone (aka “enone”) where a strong base (NaOCH3 in this case) is used to form the enolate: (The pKa of malonic esters is usually around 13, so this is quite a favorable acid-base reaction)
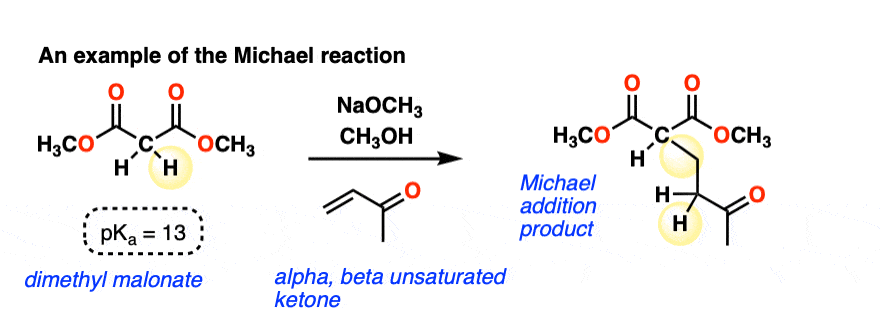
A new C-C single bond is formed, and so is a new C-H bond on the alpha-carbon of the alpha, beta unsaturated ketone. The C-H bond on the alpha carbon of the malonic ester is broken, and so is the C-C pi bond.
The nucleophile can also add to alpha,beta-unsaturated nitriles, such as in this example:
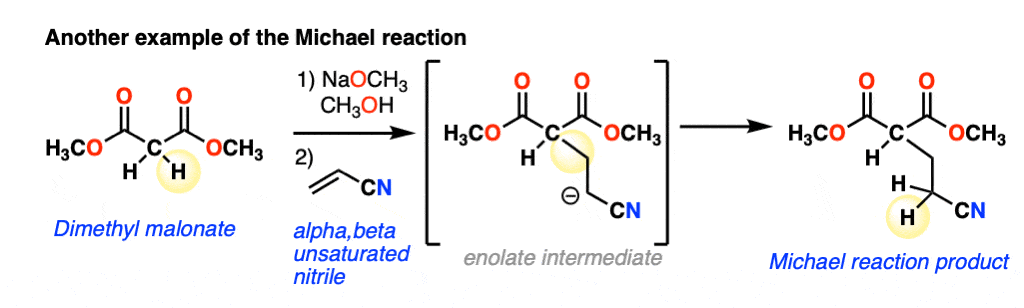
The scope goes beyond alpha,beta unsaturated ketones and alpha,beta unsaturated nitriles to include alpha,beta unsaturated esters, nitro compounds, sulfonic esters and more. Because it can get lengthy to write these names out, the term “Michael acceptor” is often used to refer to alkenes [Note 1] attached to one or more electron withdrawing groups that can act as pi-acceptors.(See article: Exploring Resonance: Pi Acceptors)
Here is an example of the Michael reaction between a ketone enolate (made through selective deprotonation of the less-substituted alpha carbon with the bulky base lithium di-isopropyl amide (LDA) – see article – Kinetic vs Thermodynamic Enolates) and an alpha,beta unsaturated ketone.
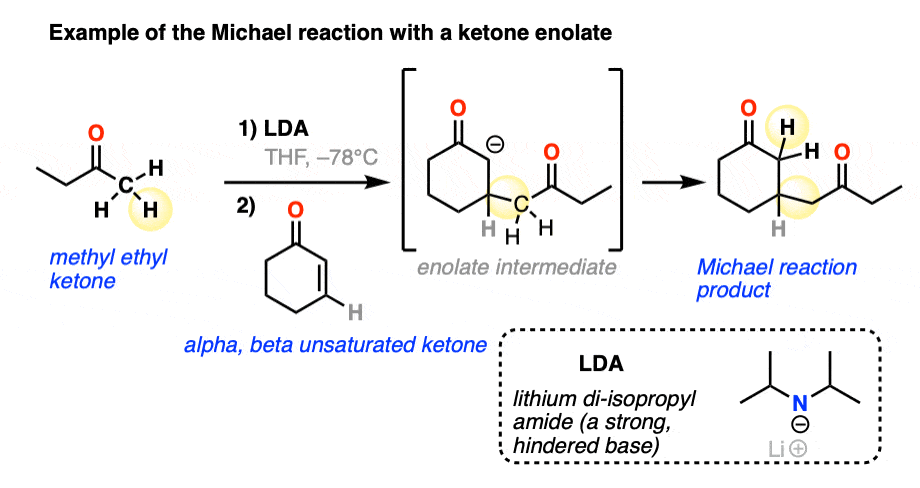
With those three examples in mind, let’s draw a general scheme!
2. General Pattern of the Michael Reaction
The general pattern for a Michael reaction looks like this:
- An enolate (formed through treatment of a ketone, ester, nitrile or similar species with base) adds to the beta-carbon of an electrophilic alkene such as an alpha, beta-unsaturated ketone, nitrile or nitro compound (“Michael acceptor”)
- …giving a new C-C single bond and breaking a C-C pi bond
- …resulting in a new enolate, which is then protonated to give the final product.
The electron withdrawing group attached to the alkene is generally a carbonyl compound such as a ketone or ester, but can also be a nitrile, nitro or sulfonyl, among others.
The driving force for the Michael is the exchange of a relatively weak C-C pi bond (about 60 kcal/mol) for a relatively strong C-C single bond (about 80 kcal/mol).
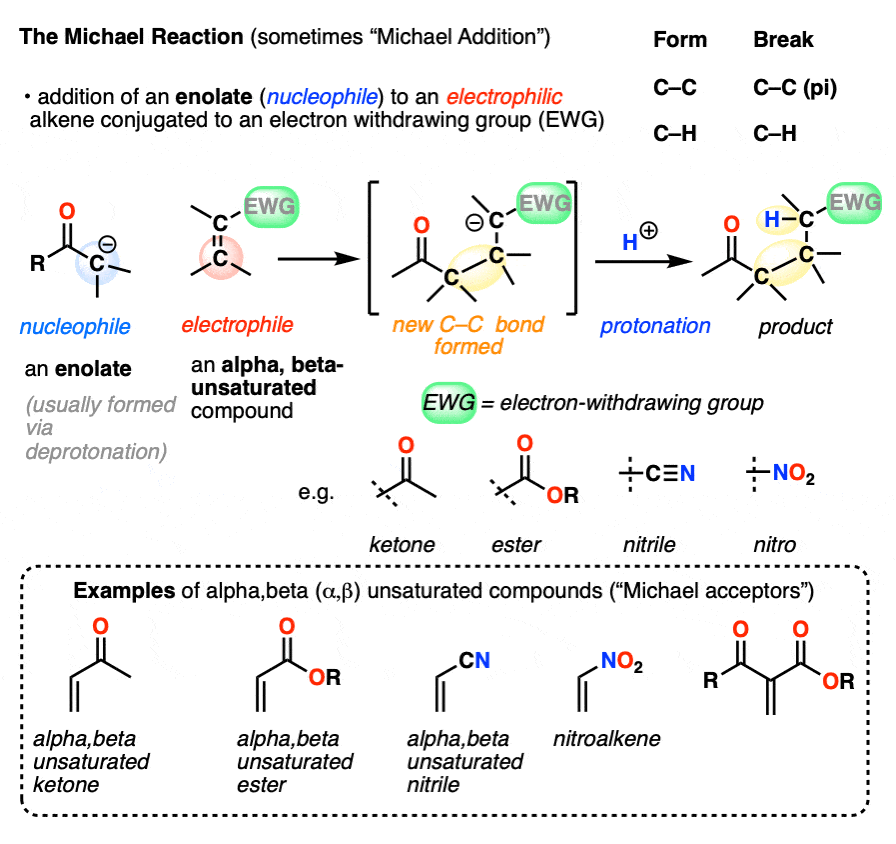
A typical product of a Michael reaction will have two carbonyl groups separated by 3 carbons. (A general name for this is a “1,5-dicarbonyl”, although in some cases a nitro-, cyano- or sulfonyl group stands in for one or more of the carbonyl carbons. )
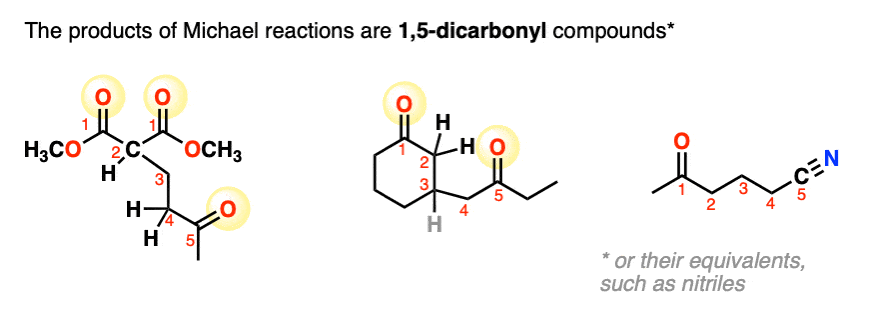
3. Mechanism of the Michael Reaction
The Michael reaction involves just three key steps:
- deprotonation of the starting material to give an enolate
- conjugate addition (1,4-addition) of the enolate to an electrophilic alkene, and
- protonation of the resulting enolate
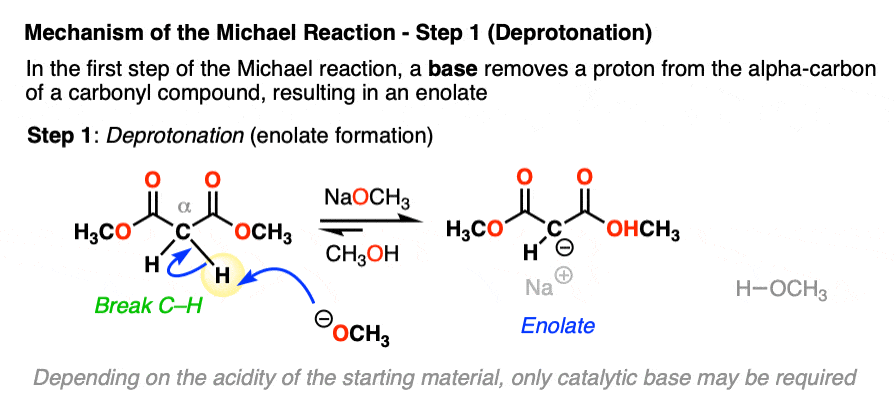
Let’s look at the first step with a malonic ester like in the first example of this article.
In the first step, sodium methoxide (NaOCH3) removes a proton from the alpha carbon, resulting in a new enolate.
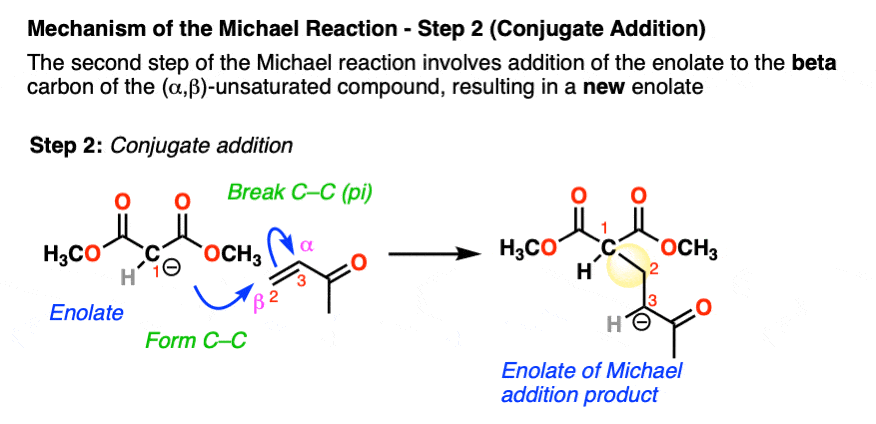
Enolates are excellent nucleophiles. In the second step, the enolate adds to the beta carbon of the alpha, beta unsaturated ketone, which results in a new enolate. This mechanistic step is often known as conjugate addition or (sometimes) 1,4-addition.
The final step is protonation of the resulting enolate to give the neutral Michael addition product.
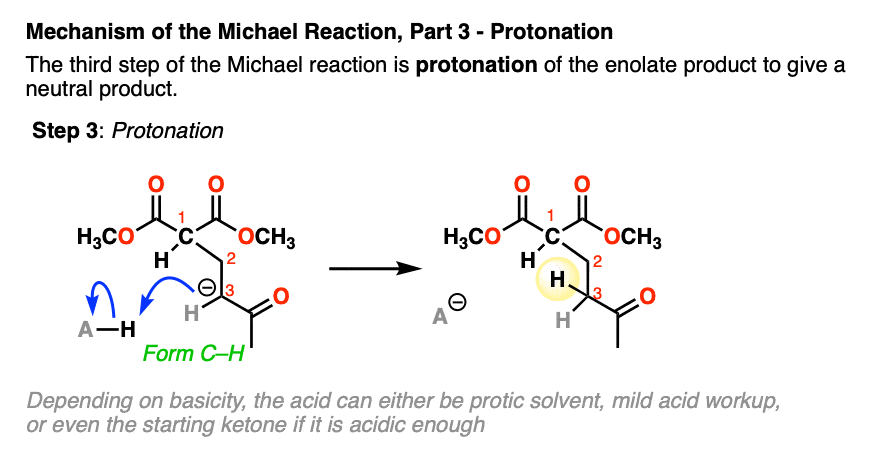
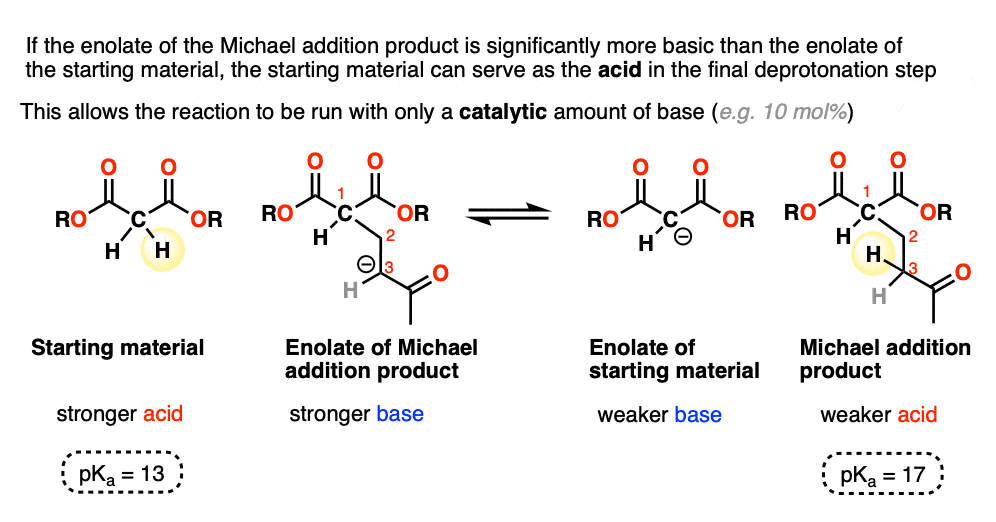
When the enolate of the product is significantly more basic than the enolate of the starting material, the reaction can be run using a catalytic amount of base.
4. Why Don’t Nucleophiles Add To “Normal” Alkenes?
If you step back for a second, Michael reactions might seem a little unusual.
After all, back in the day, you likely did an entire Org 1 unit on reactions of alkenes – and I’ll wager that not one of those examples involved adding a nucleophile to an alkene. (Most of the alkene reactions we saw involved reactions of electrophiles with alkenes, such as acids, halogens, m-CPBA, OsO4, O3 and so on)
So why is addition to an alkene favorable when the alkene is adjacent to a ketone, ester, or other electr0n-withdrawing group, but unfavorable for “normal” alkenes like, say, cyclohexene?
It might be helpful to draw out the important resonance forms of an alpha,beta-unsaturated ketone along with its resonance hybrid.
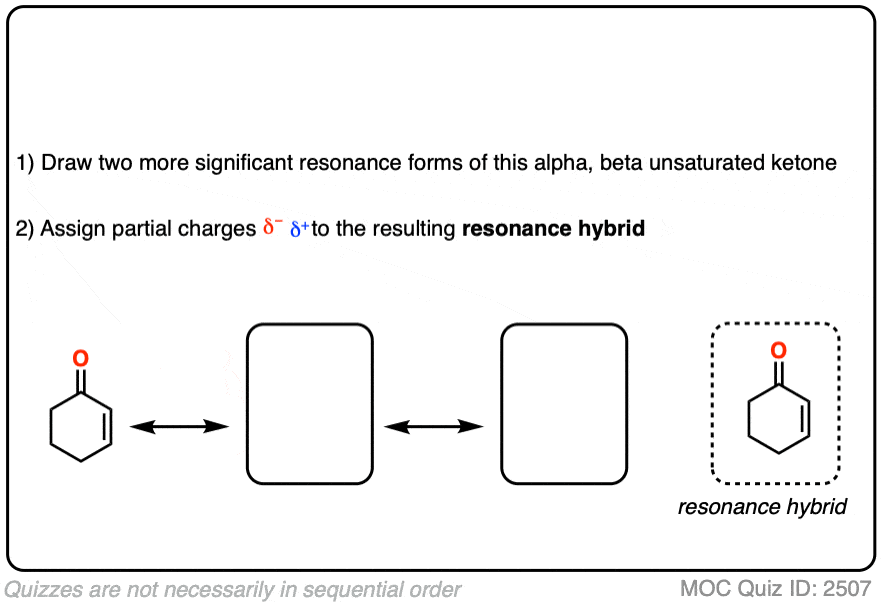 Click to Flip
Click to Flip
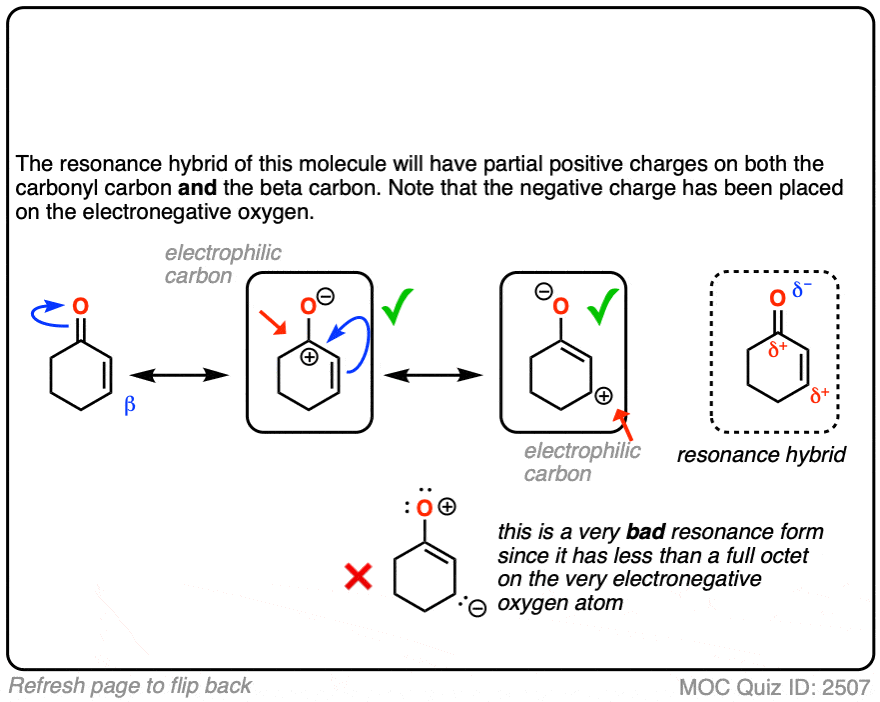
When you draw out the key resonance form, you’ll notice there is a positive charge on the beta carbon and a negative charge on the oxygen. The beta-carbon is electrophilic, in other words, and the carbonyl oxygen acts as an electron “sink” for the electrons in the C-C pi bond.
The reactivity of alpha,beta unsaturated systems makes sense when you think about the stability of the negative charge that results from addition of a nucleophile to the beta carbon. (See post – 7 Factors That Stabilize Negative Charge)
Addition of a nucleophile to an alkene results in a new negative charge on carbon (also known as a “carbanion”)
Addition of nucleophiles to ordinary alkenes is strongly disfavored since the resulting carbanion is an extremely strong base (recall that the pKa of alkanes is >50 or so). [Note 2 ]
On the other hand, when a ketone, nitrile, ester or other pi-acceptor is adjacent to the alkene, the resulting negative charge can be delocalized through resonance to oxygen or nitrogen.
Being more electronegative than carbon, these atoms are better able to stabilize negative charge. That’s why a typical ketone has a pKa of about 17 or so (30 orders of magnitude more acidic than an ordinary alkane!)
Or to state things more briefly, addition to these alkenes results in an enolate, and enolates are considerably more stable than ordinary carbanions since negative charge can be transferred to oxygen.
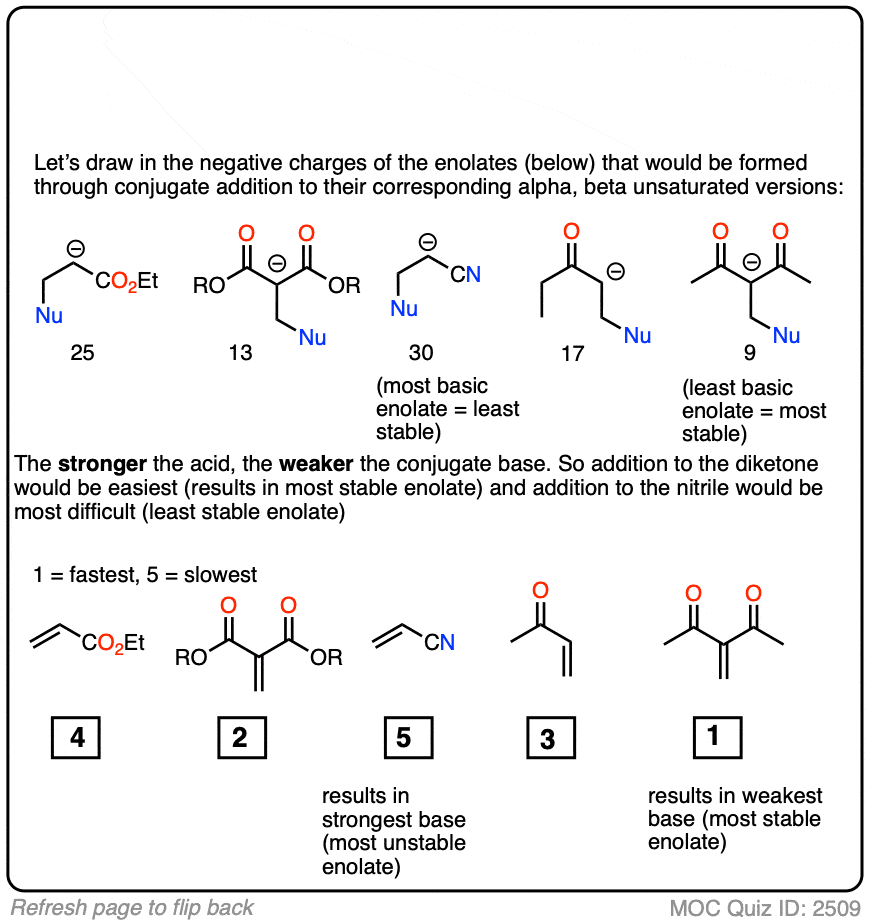
In fact, the ease of conjugate addition correlates extremely well with the stability of the resulting enolate. Try this exercise, for instance:
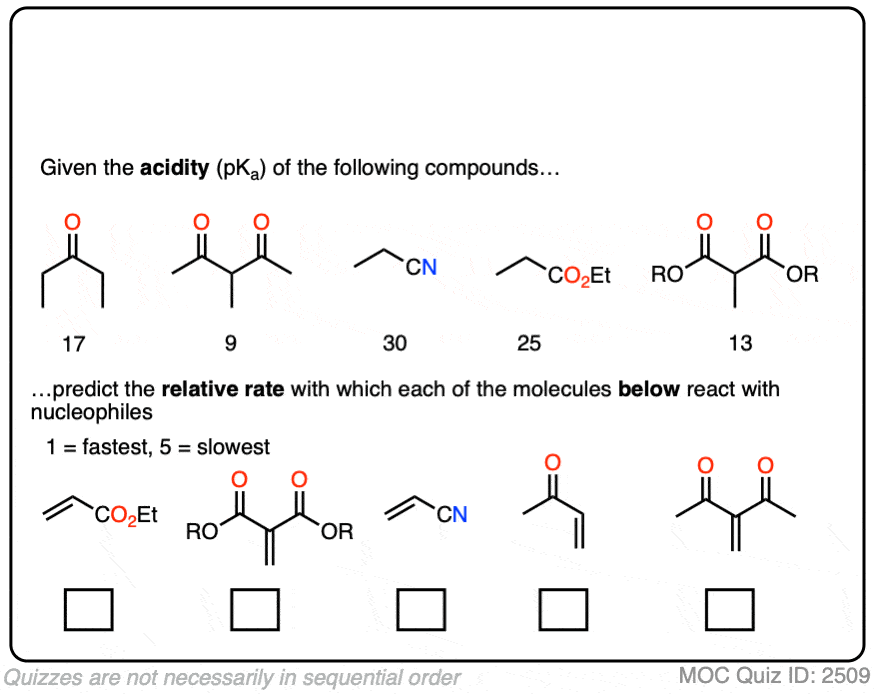 Click to Flip
Click to Flip
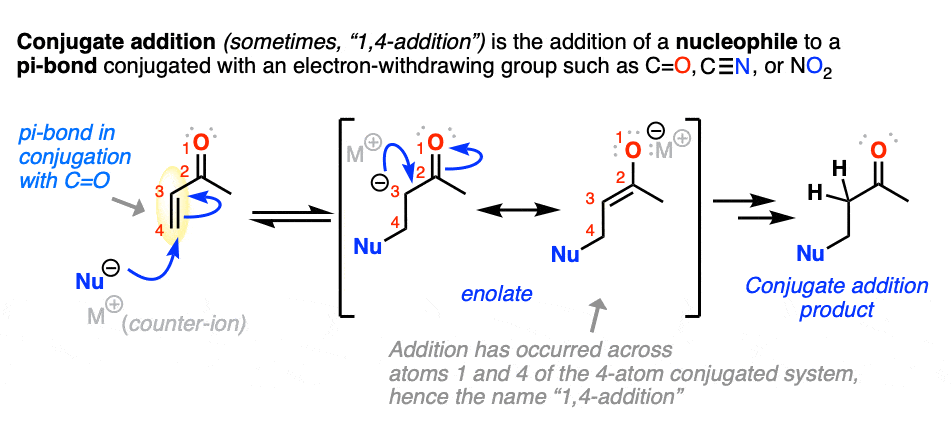
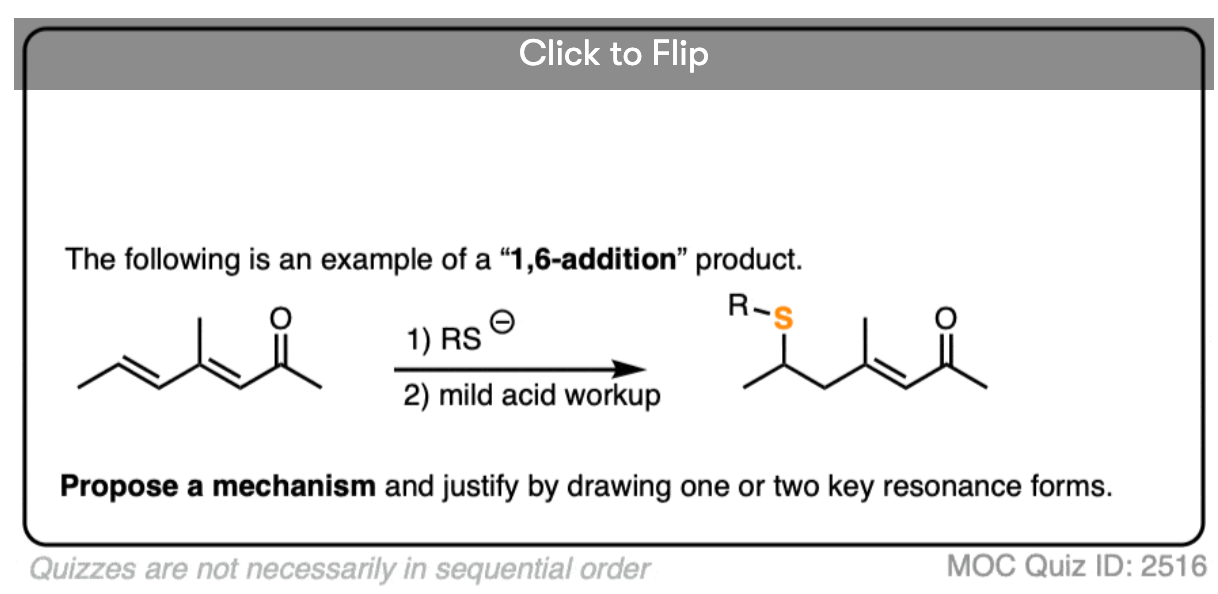
5. Conjugate Addition (“1,4-Addition”)
Michael reactions are part of a more general class of reactions known as conjugate addition.
Conjugate addition is the addition of a nucleophile to an alkene that is in conjugation with a carbonyl (C=O), cyano (CN), nitro (NO2) or other functional group that can act as a “pi acceptor”.
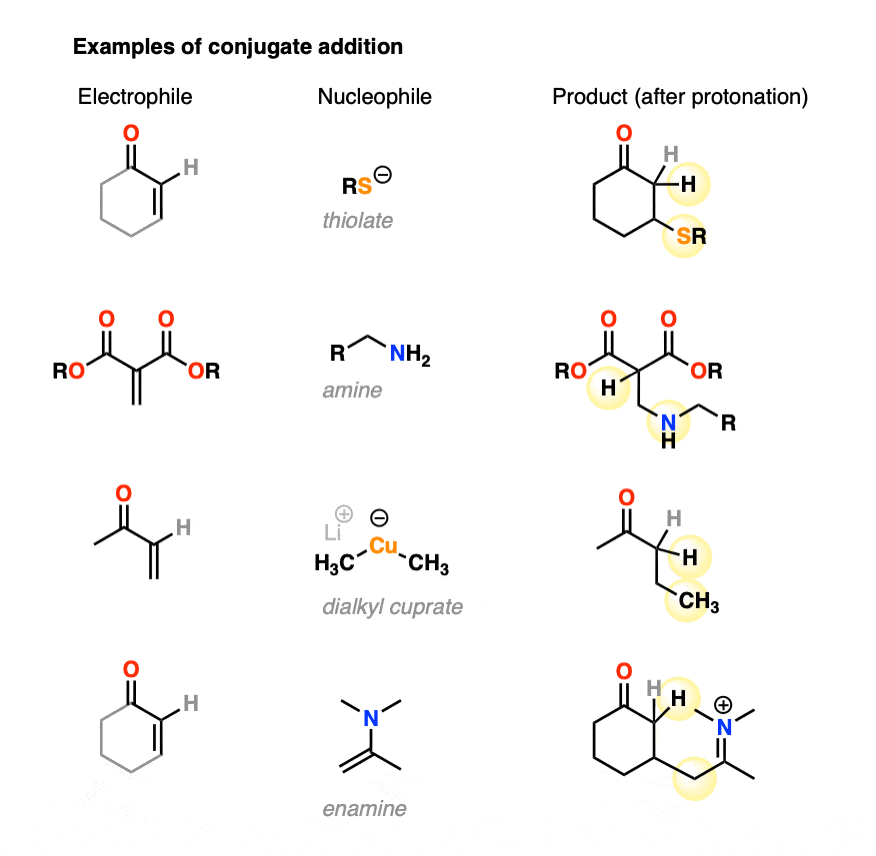
Conjugate addition can occur with a great deal of different nucleophiles, such as amines, thiolates (RS(-), enamines, and dialkylcuprates, among many others. Here are some representative examples.
6. 1,2-Addition Versus 1,4-Addition
All right. So far, so good. I regret to inform you that the time has arrived to make our discussion more complicated.
A Michael acceptor such as an alpha,beta-unsaturated ketone does not have just one electrophilic carbon. It actually has two: the beta-carbon (which we’ve been focusing on here) in addition to the carbonyl carbon.
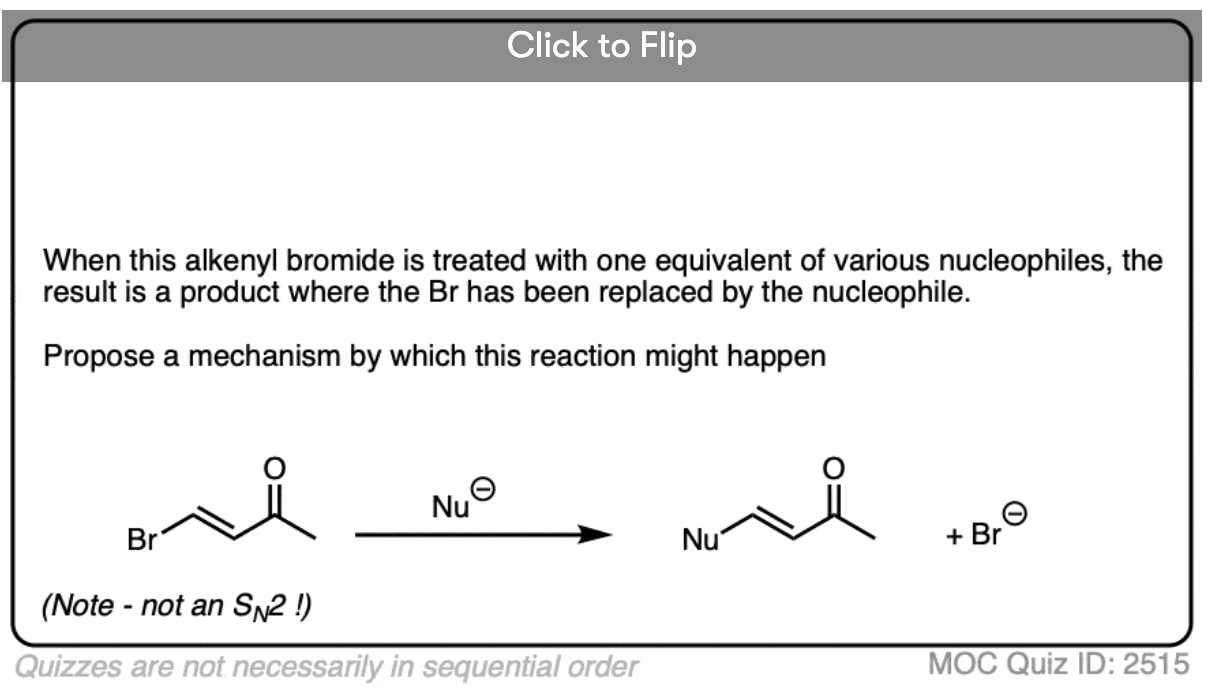
Conjugate addition is sometimes called, “1,4-addition” to contrast it with “1,2-addition”, which is another name for nucleophilic addition to the carbonyl (C=O) (See article: Nucleophilic Addition to Carbonyls)
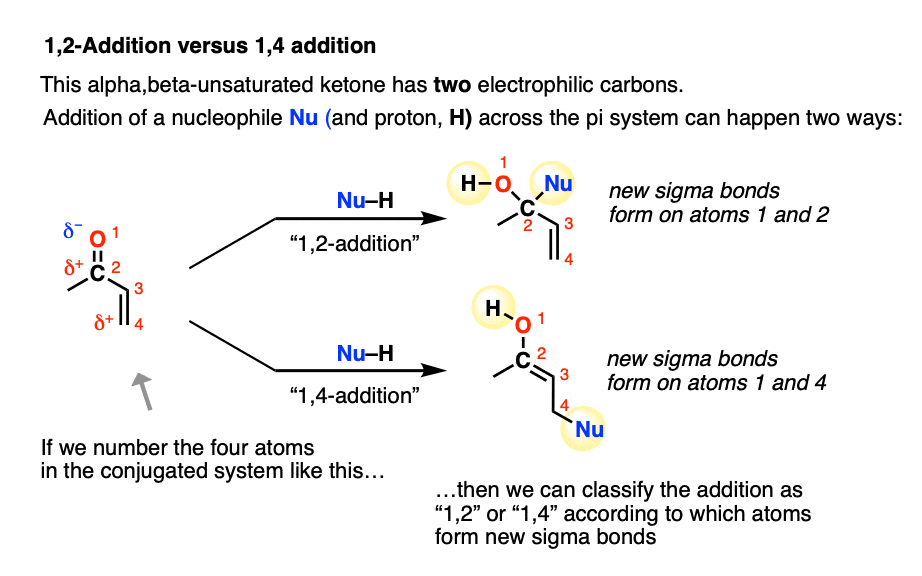
In molecules like enones, both 1,2- and 1,4- addition can occur. (Michael acceptors with less electrophilic carboxylic acid derivatives such as amides and nitriles will have less competition from the 1,2-pathway).
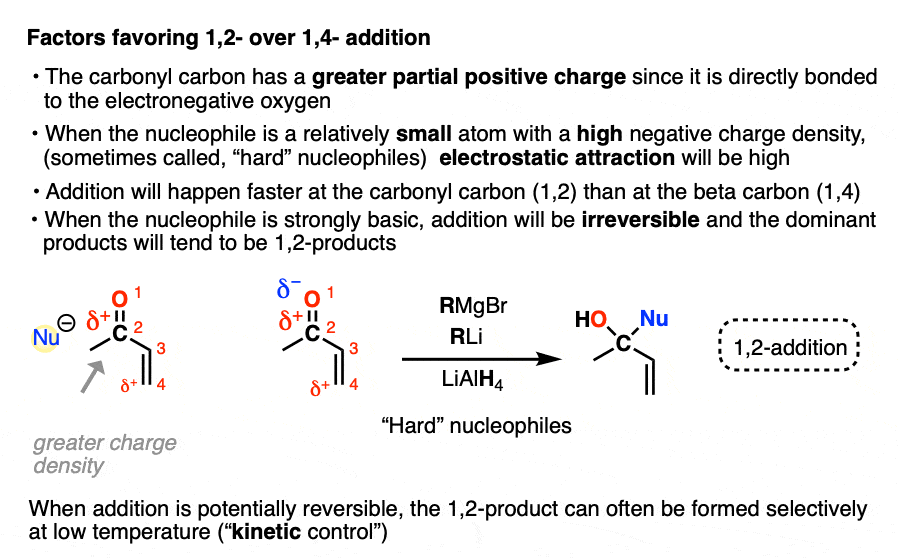
- “1,2-addition” tends to be faster due to the fact that the carbonyl carbon is directly attached to oxygen and has a greater partial positive charge than the beta carbon. When addition is irreversible, as it is with many strongly basic nucleophiles (or through keeping the reaction at low temperature through kinetic control) 1,2-products will often dominate.
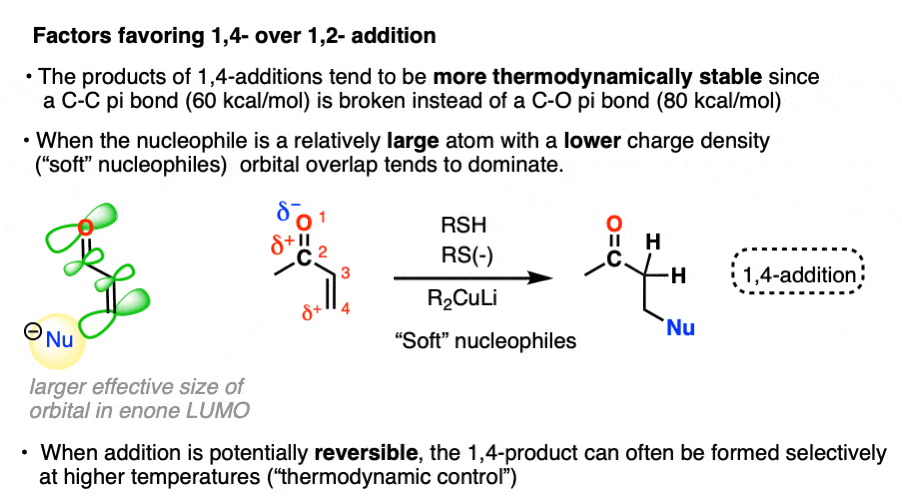
- “1,4-addition” products tend to be more thermodynamically stable since they involve breaking a C-C pi bond (60 kcal/mol) instead of a C-O pi bond (80 kcal/mol). When the reaction is reversible, or heated such that an equilibrium is established (thermodynamic control) 1,4-products will often dominate.
Unlike our previous discussions of kinetic vs. thermodynamic control, however, (See article – Kinetic and Thermodynamic Control) temperature isn’t everything. The identity of the nucleophile plays a key role.
A classic example of contrasting reactivity is found through comparing the reactions of Grignard reagents (RMgBr) and organocuprates (Gilman reagents, R2CuLi) with alpha, beta unsaturated ketones (“enones”). Both are strongly basic nucleophiles that add irreversibly. However…
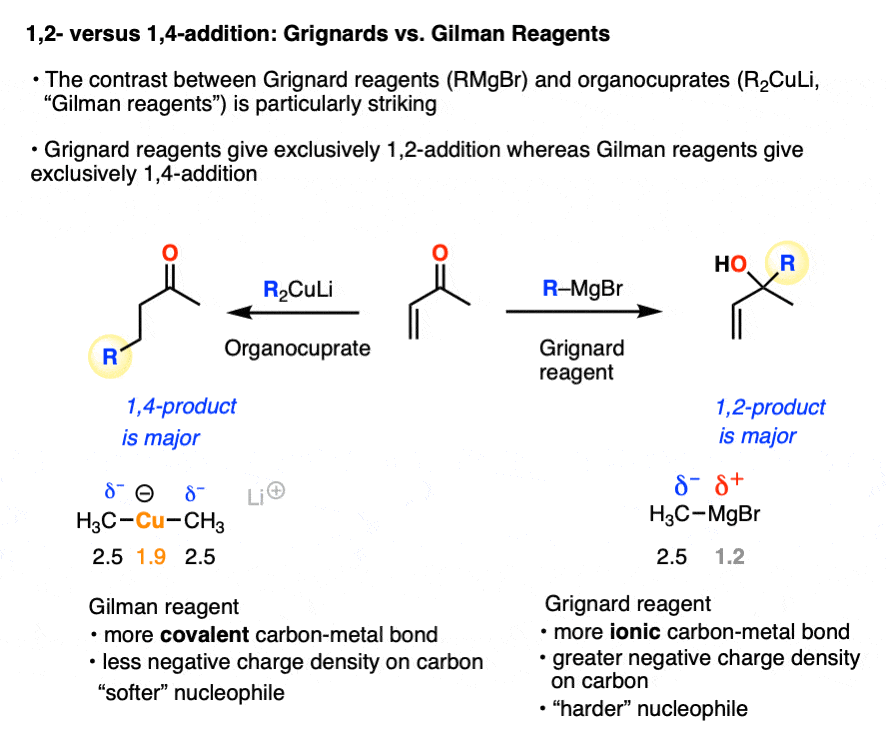
- Grignard reagents tend to preferentially give 1,2-addition (as do organolithium reagents and hydrides such as LiAlH4 )
- Organocuprates tend to preferentially give 1,4-addition (as do enolates, enolates, thiols, amines, and enamines)
So what’s going on? That is a great question.
A working model for explaining these divergent patterns is known as Hard Soft Acid-Base Theory (HSAB). In HSAB, “hard” nucleophiles tend to react with “hard” electrophiles, and “soft” nucleophiles tend to react with “soft” electrophiles. [Note 3]
[For more on HSAB and a summary of the factors that promote 1,2- versus 1,4-addition, see Note 4]
7. Intramolecular Michael Reactions
Rule 34 of organic chemistry is, “If a reaction exists, there is an intramolecular variant of it”.
The Michael reaction and conjugate addition are no different.
The key thing about intramolecular reactions, as you might see me say many times on this site, is that from an instructors’ point of view, they involve no new concepts but offer the element of surprise. As such, they make for great exam questions.
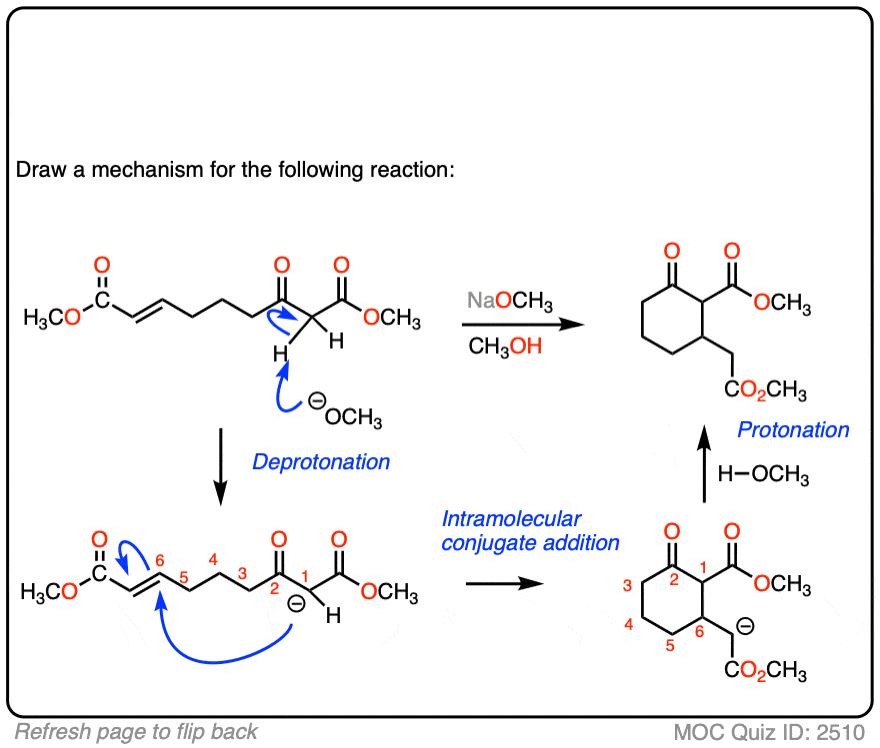
See if you can draw the mechanism for this intramolecular Michael reaction. Remember to number your carbons, and draw the ugly version first if it is not immediately clear, just so you can get the bond forming/ bond breaking right. You will always have time to draw a prettier version later.
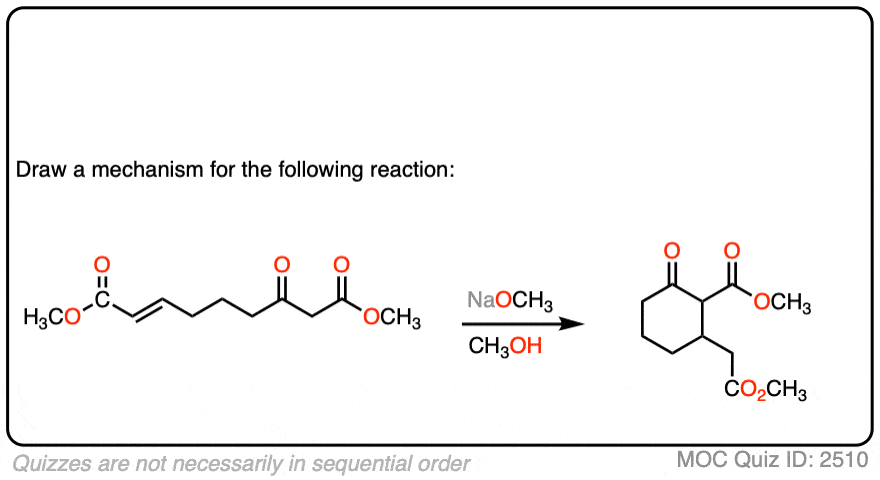 Click to Flip
Click to Flip
Here is another test. In this case, draw the product of this intramolecular Michael reaction.
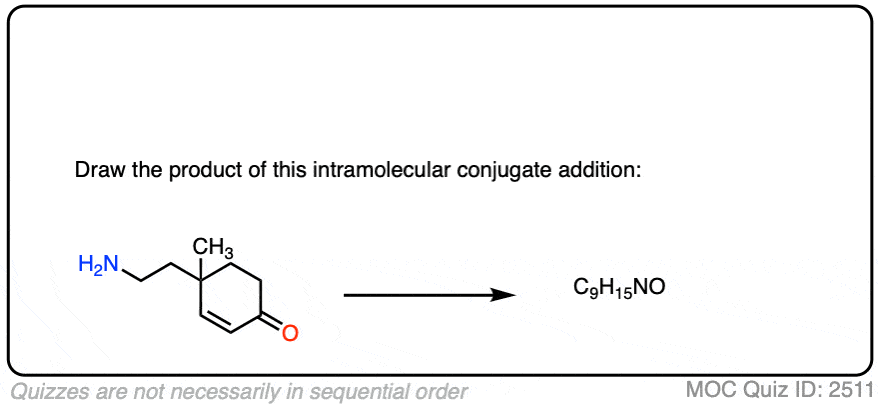 Click to Flip
Click to Flip
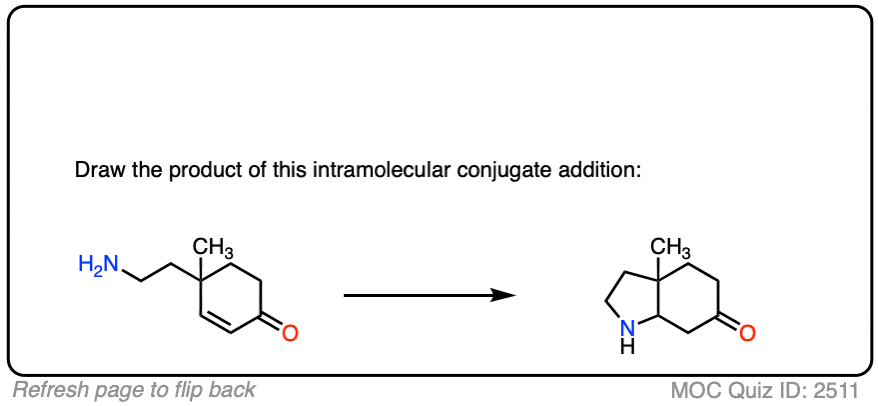
8. Michael Additions and Synthesis: Working Backwards
I’m not sure what rule number this one is, but any time you learn a new reaction, you can be expected to apply it in a synthesis problem. This means being able to work backwards from the product to plausible starting materials.
See if you can disconnect these Michael addition / conjugate addition products to get back appropriate starting materials.
Hint: recall that the key pattern in a Michael reaction is formation of 1,5-dicarbonyl.
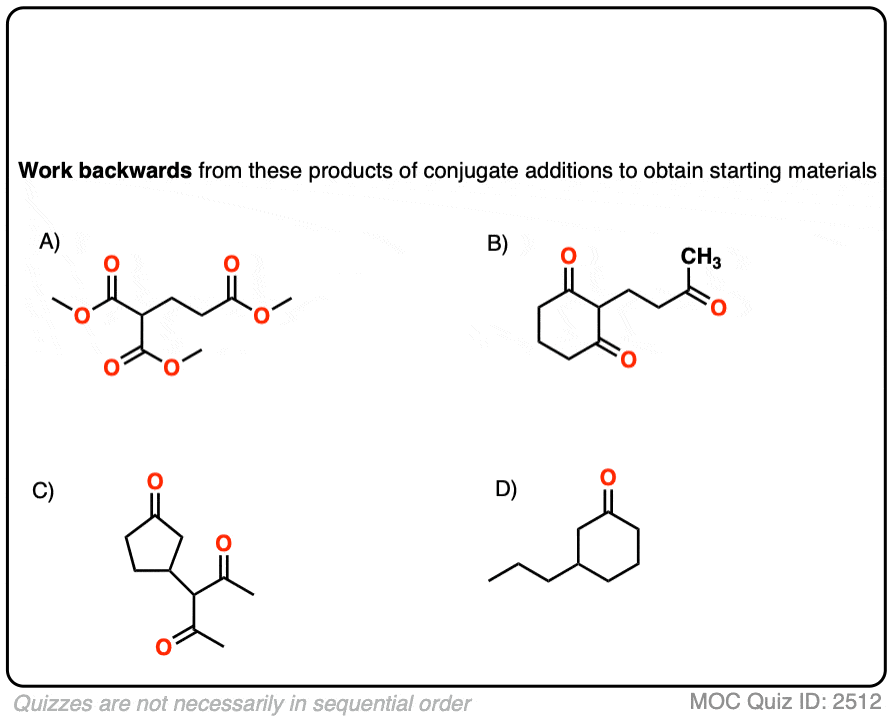 Click to Flip
Click to Flip
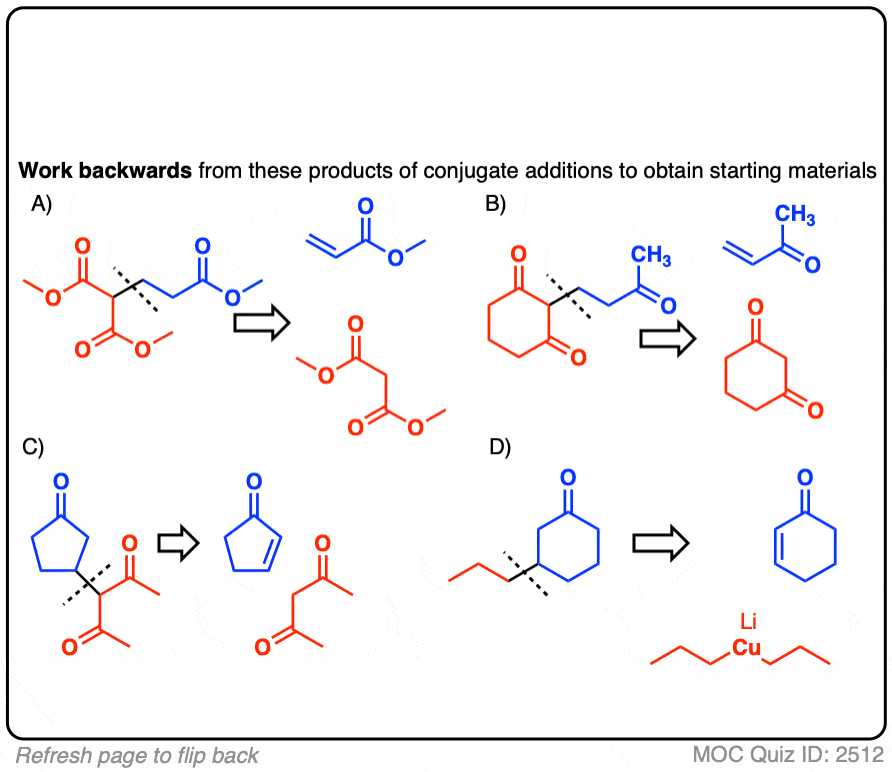
There may be more than one correct answer; the more acidic starting material is shown in each case.
9. Summary
- We’ve seen that the key step in the Michael addition is the conjugate addition of an enolate to an electrophilic alkene.
- The reaction proceeds in three steps: 1) deprotonation 2) conjugate addition, and 3) protonation
- Alkenes in conjugation with a pi-acceptor such as a carbonyl, nitrile, nitro, ester (and others) have an important resonance form where the beta-carbon bears a positive charge. Unlike ordinary alkenes, nucleophilic addition to this carbon is possible because the pi-acceptor is able to accept a lone pair from a pi bond and stabilize the negative charge on a more electronegative atom than carbon.
- Conjugate addition can happen with a variety of nucleophiles such as enolates, organocuprates, thiols, amines, enamines and more.
- When both 1,2- and 1,4- addition are reversible, equilibrium tends to favor formation of the 1,4-product due to breakage of the weaker C-C (pi) bond.
- Strongly basic, ionic nucleophiles that add irreversibly (such as Grignard and organolithium reagents) tend to give products of 1,2-addition selectively over 1,4-addition.
- Organocuprates tend to give 1,4-addition products exclusively, as do thiols, phosphines, enamines and others.
One key application of the Michael reaction is in the Robinson Annulation, which involves Michael addition followed by aldol condensation. (See article: The Robinson Annulation)
Notes
Note 1. Although I wrote “alkenes” here, Michael-type reactions can also occur with electrophilic alkynes.
Note 2. If this sounds like we’re boiling this down to a fancy acid-base reaction… you’re not wrong! A lot of reactions can be judged on whether they are favorable or unfavorable depending on whether or not the product is more unstable (more basic) than the starting material. It’s handy to think of “basicity” as just a fancy name for “unstable lone pair of electrons”.
Note 3. Hard-Soft Acid Base theory is a distillation of the Klopman-Salem equation of reactivity which has two key terms: 1) a contribution from electrostatic attraction, and 2) a contribution from orbital overlap, which is highest when the HOMO and LUMO are close together in energy. (The Wikipedia article on the Klopman-Salem is very bad – see Fleming’s classic Frontier Orbitals And Organic Chemical Reactions for a much better discussion)
Note 4. Hard-Soft Acid-Base Theory (HSAB)
In one mode of reactivity (“hard-hard interactions”), the reaction products are driven by the electrostatic attraction of highly electron-rich ( δ–) nucleophiles toward highly electropositive ( δ+ )electrophiles. This is most effective for relatively small, non-polarizable atoms with high charge densities, which are termed “hard” in this model (Like a charged little billiard ball, the proton H+ is the prototypical “hard” electrophile).
This allows us to rationalize the preference for 1,2-addition of Grignards and organolithiums as an interaction between a “hard” electrophile and a “hard” nucleophile.
- the carbonyl carbon is more electropositive (“harder”) than the beta carbon since it’s directly attached to electronegative oxygen
- Grignard and organolithium reagents have a more ionic carbon-metal bond due to the lower electronegativity of Li (1.0) and Mg (1.2) relative to Cu (1.9). This means carbon will have greater electron density (more δ– = “harder”) than it does in organocuprates, where the C-Cu bond is more covalent (electronegativity of C is 2.5)
For electrostatic reasons, 1,2-addition also tends to be faster than 1,4-addition.
In the other mode of reactivity (“soft-soft interactions”) the product distribution is driven not so much by by orbital overlap between the lone pair of the nucleophile and the lowest-unoccupied molecular orbital (LUMO) of the enone.
“Soft” nucleophiles and electrophiles are considered to be relatively large, polarizable atoms with low charge densities – like a big squishy Nerf ball of charge that can be easily squeezed and deformed. Prototypical “soft” nucleophiles include alkenes, thiols, and iodide. “Soft” electrophiles include electron-deficient alkenes like enones. Pericyclic reactions like the Diels-Alder are entirely based on “soft-soft” interactions.
For complex reasons, not all atoms of the LUMO have the same effective size. [Note 5] In the LUMO of the enone, the beta carbon has a greater effective size than the carbonyl carbon. In the absence of charge effects, the reaction will occur at the site where the greatest overlap can occur between the HOMO of the nucleophile the LUMO of the electrophile.
Note 5. In Huckel Molecular Orbital theory, each pi molecular orbital is described as a linear combination of the 2p atomic orbitals at carbon, with coefficients c relating the size of the contribution at each carbon. [every carbon is not equal]
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
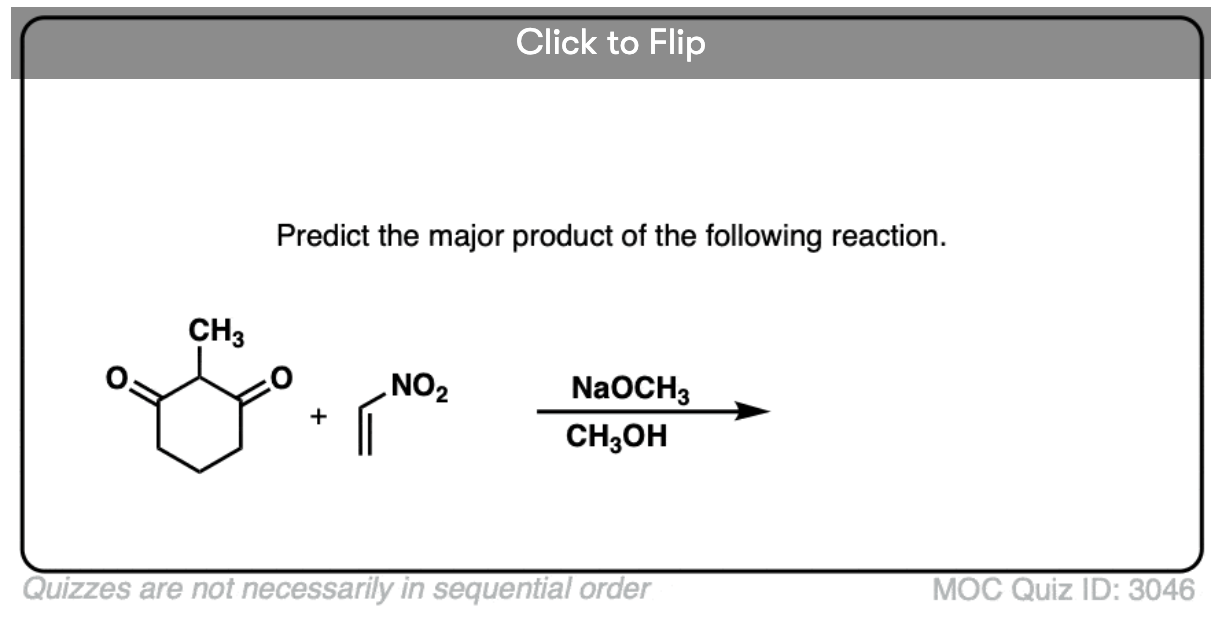
Become a MOC member to see the clickable quiz with answers on the back.
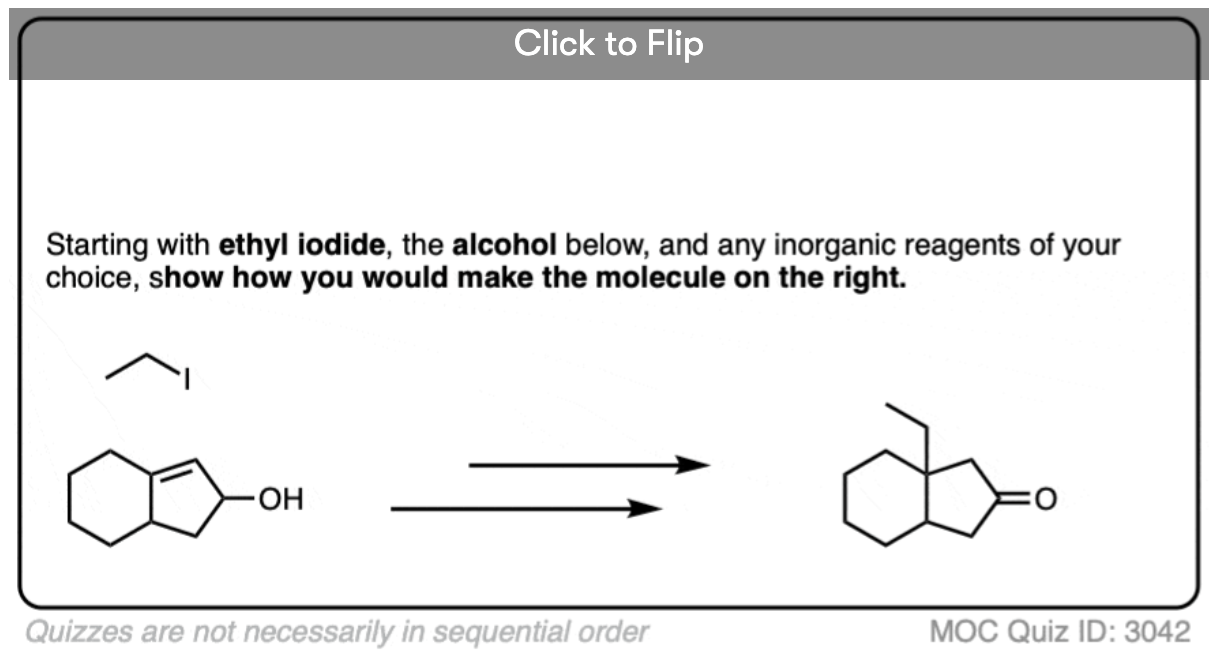
Become a MOC member to see the clickable quiz with answers on the back.
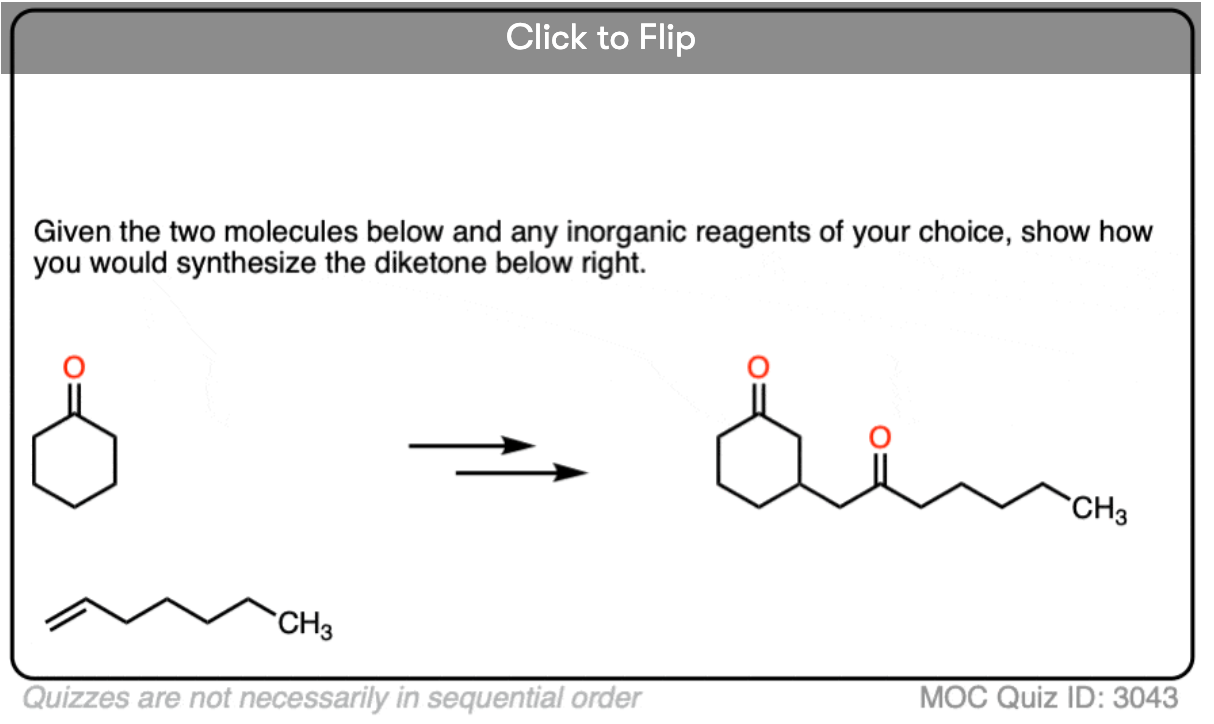
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Ueber die Addition von Natriumacetessig‐ und Natriummalonsäureäthern zu den Aethern ungesättigter Säuren
A. Michael, J. Prakt. Chem. 1887 35 (1), 349-356
DOI: 10.1002/prac.18870350136
The original paper by Arthur Michael on the addition of carbon nucleophiles to a,b-unsaturated systems. Note that even though Michael was an American chemist at Tufts College (see the very end of the paper), this was still written in German and published in a German journal since German chemistry journals were the best at the time. - The Michael Reaction
Bergmann, E. D.; Ginsburg, D.; Pappo, R. React. 1959, 10, 179
DOI: 10.1002/0471264180.or010.03
Classic review on the Michael Reaction. This also has detailed experimental procedures towards the end, similar to what you might find in Organic Syntheses. - (R)-3-PHENYLCYCLOHEXANONE
Tamio Hayashi, Makoto Takahashi, Yoshiaki Takaya, and Masamichi Ogasawara
Org. Synth. 2002, 79, 84
DOI: 10.15227/orgsyn.079.0084
If a chiral Lewis acid is used, then asymmetric 1,4-additions are also possible,.The theory underlying why certain nucleophiles or Lewis acids prefer 1,4 or 1,2 addition is “Hard Soft Acid Base Theory”, or HSAB. This was developed by R. G. Pearson in the 1960’s and is very useful, since it allows chemists to quickly and qualitatively determine how a molecule or ion will react based on its polarizability. - Hard and Soft Acids and Bases
Ralph G. Pearson
Journal of the American Chemical Society 1963, 85 (22), 3533-3539
DOI: 10.1021/ja00905a001 - Acids and Bases
Ralph G. Pearson
Science 14 Jan 1966: Vol. 151, Issue 3707, pp. 172-177
DOI: 10.1126/science.151.3707.172 - Hard and soft acids and bases, HSAB, part 1: Fundamental principles
Ralph G. Pearson
Journal of Chemical Education 1968, 45 (9), 581
DOI: 10.1021/ed045p581 - Hard and soft acids and bases, HSAB, part II: Underlying theories
Ralph G. Pearson
Journal of Chemical Education 1968, 45 (10), 643
DOI: 10.1021/ed045p643 - Hard soft acids bases (HSAB) principle and organic chemistry
Tse-Lok Ho
Chemical Reviews 1975, 75 (1), 1-20
DOI: 10.1021/cr60293a001 - Chemoselectivity of organometallic reactions : A HSAB appraisal
Tse-Lok Ho
Tetrahedron 1985, 41 (1), 3-86
DOI: 10.1016/S0040-4020(01)83470-0
References #6 and #7 cover the specific application of HSAB theory to organic chemistry, showing how certain reaction mechanisms can be explained on the basis of this theory. - An ab initio study resulting in a greater understanding of the HSAB principle
Pratim K. Chattaraj and Paul v. R. Schleyer
Journal of the American Chemical Society 1994, 116 (3), 1067-1071
DOI: 10.1021/ja00082a031 - Farewell to the HSAB Treatment of Ambident Reactivity
Prof. Dr. Herbert Mayr, Dr. Martin Breugst, Dr. Armin R. Ofial
Angewandte Chemie International Edition in English 2011, 50 (29), 6470-6505
Dissenting opinion on HSAB by Prof. Mayr: “The prediction of ambident reactivity by the concept of hard and soft acids and bases fails in the majority of cases and has to be abandoned. In this Review an alternative approach is presented to rationalize the regioselectivities of ambident systems which differentiates between kinetic and thermodynamic product control as well as between reactions proceeding with and without activation energy. The predictive power of qualitative Marcus theory is demonstrated.” - The asymmetric Michael addition reactions using chiral imines
Jean d’Angelo, Didier Desmaële, Françoise Dumas, André Guingant, Vedejs, E.
Tetrahedron Asymmetry 1992, 3 (4), 459
DOI: 10.1016/S0957-4166(00)80251-7
Michael reactions can also be asymmetric; if you have a chiral imine instead of a carbonyl, then stereoselective 1,4-addition is possible. - The Enantioselective Organocatalytic 1,4-Addition of Electron-Rich Benzenes to α,β-Unsaturated Aldehydes
Nick A. Paras and and David W. C. MacMillan
Journal of the American Chemical Society 2002, 124 (27), 7894-7895
DOI: 10.1021/ja025981p
This is a modern twist on the previous reference by Prof. MacMillan, who has made important contributions to organic chemistry. This paper shows that asymmetric Michael additions can be done through chiral imine intermediates by generating the chiral imine in situ using a chiral amine catalyst. The concept is simple and elegant.
I really enjoyed this site🙏