Home / The Pi Molecular Orbitals of Cyclobutadiene
Aromaticity
The Pi Molecular Orbitals of Cyclobutadiene
Last updated: October 27th, 2022 |
Cyclobutadiene: Molecular Orbital Diagram, Antiaromaticity, and Structure
Previously, we’ve seen what the molecular orbitals of benzene look like, and that the fact that they are partially duplexed (or to use the proper nomenclature, “degenerate“) helps to explain benzene’s unusual stability.
Let’s flip the coin. What about cyclobutadiene, a molecule we usually class as antiaromatic. Why is it so unusually unstable?
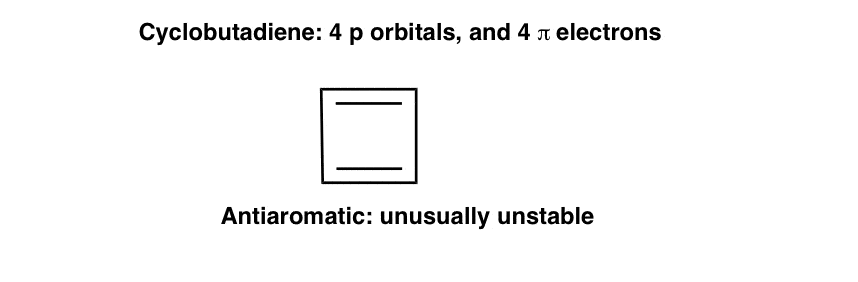
Again, examining the pi molecular orbitals will give us some useful clues.
Notably, in cyclobutadiene, the highest-occupied molecular orbitals are unpaired electrons of equal energy.
Table of Contents
- Building Up The Molecular Orbital Diagram Of Cyclobutadiene: The Lowest-Energy Molecular Orbital Has Zero Nodal Planes
- The Highest-Energy Molecular Orbital Has Two Nodal Planes
- The Two Intermediate pi Molecular Orbitals Each Have One Nodal Plane (two different ways)
- The Molecular Orbital Diagram Of Cyclobutadiene Reveals Why Cyclobutadiene Is Extremely Unstable: It Has Unpaired Electrons Of Equal Energy
- Summary: The Molecular Orbital Diagram of Cyclobutadiene
- Notes
- (Advanced) References and Further Reading
1. Building Up The Molecular Orbital Diagram Of Cyclobutadiene: The Lowest-Energy Molecular Orbital Has Zero Nodal Planes
Today, let’s build up the orbitals of cyclobutadiene using the principles we’ve discussed in previous posts [e.g. see this post on butadiene] and see if we can gain some useful insights.
Cyclobutadiene has a pi system comprised of 4 individual atomic p orbitals and thus should have a total of 4 pi molecular orbitals.
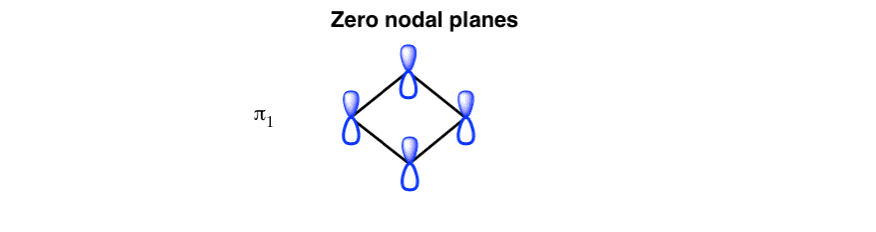
The lowest-energy molecular orbital: zero nodal planes
Following our “apartment building” analogy from last time, the lowest-energy molecular orbital (the “ground floor” of cyclobutadiene, if you will) should have all phases of the p-orbitals aligned and zero nodal planes, like this:
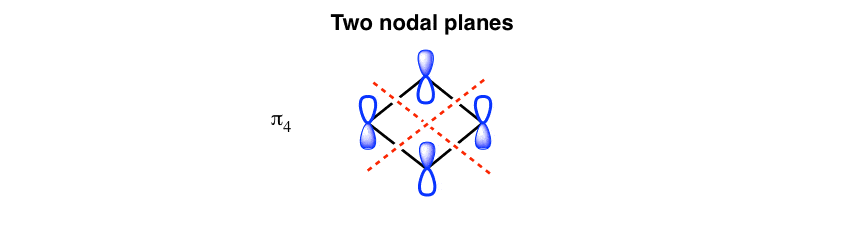
2. The Highest-Energy Molecular Orbital Has Two Nodal Planes
Conversely, the highest-energy pi orbitals (the “penthouse”) will have all phases alternating, and thus have two nodal planes. (As we said last time, the “penthouse” is not exactly desirable real estate for electrons)
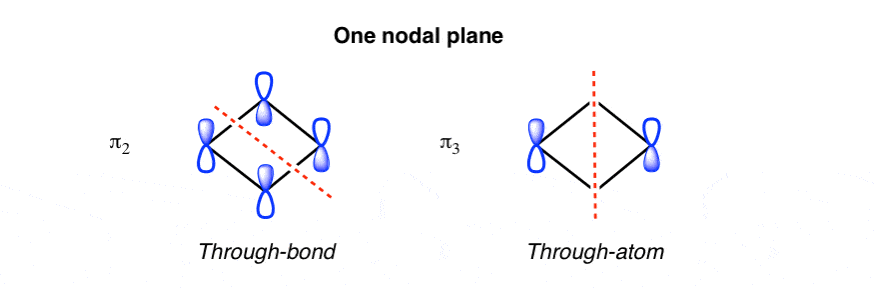
3. The Two Intermediate pi Molecular Orbitals Each Have One Nodal Plane (two different ways)
That leaves us with the intermediate pi orbitals, which each have a single nodal plane. As with benzene, there are two ways to place a single nodal plane on cyclobutadiene, either through the bonds, or through the atoms:
That gives us our four molecular orbitals. Now lets populate them with the “tenants”: the pi electrons.
4. The Molecular Orbital Diagram Of Cyclobutadiene Reveals Why Cyclobutadiene Is Extremely Unstable: It Has Unpaired Electrons Of Equal Energy
Cyclobutadiene has a total of 4 pi electrons. So ranking all the pi molecular orbitals by energy, and populating the orbitals according to Hunds rule, we get the following picture:
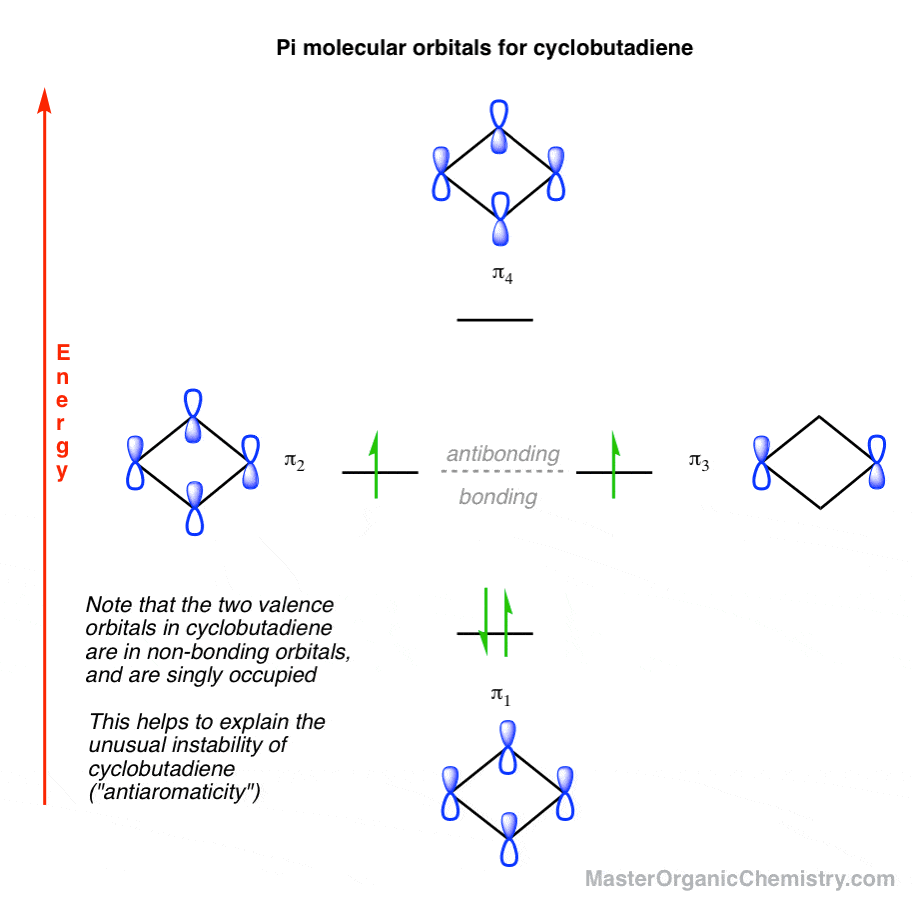
Can you see why cyclobutadiene might be unstable?
- First, the highest-occupied molecular orbitals of cyclobutadiene are non-bonding orbitals, intermediate in energy between the lowest (π1, bonding) and highest (π4, antibonding) energy orbitals. “Non-bonding” implies that filling these orbitals with electrons does not result in any stabilization of the molecule.
- Second, note that each of the non-bonding orbitals are singly occupied. Therefore this orbital picture predicts that cyclobutadiene should have a diradical nature. We’re used to thinking of free-radicals as highly reactive intermediates… so you can imagine that a species containing two free radicals is even more reactive! [Note 1 ]
5. Summary: The Molecular Orbital Diagram of Cyclobutadiene
The bottom line here is that the pi molecular orbital picture of cyclobutadiene is in agreement with our observations that cyclobutadiene is unusually unstable. (As previously noted, cyclobutadiene has only ever been isolated as a “matrix-isolated species” – that is, a species frozen in an inert gas at extremely low temperatures. Warming to a balmy –80° results in self-destruction. Note 2 )
Hopefully these two posts have helped to show that molecular orbital diagrams can provide extremely useful clues about molecular stability!
In the next post we’ll cover a very convenient short-cut that will help us quickly draw molecular orbital diagrams in seconds (yes, really!) called Frost Circles. Or, more blandly, the Polygon method.
Notes
Related Articles
Note 1. More advanced calculations, far beyond what we will discuss, predict that cyclobutadiene distorts to a rectangular shape which results in the two singly-occupied orbitals resolving into two orbitals of slightly different energy, one doubly-occupied and the other empty. The bond lengths of cyclobutadiene have been measured, confirming the rectangular shape.

Note that the pi electrons are not “delocalized” like they are in benzene.
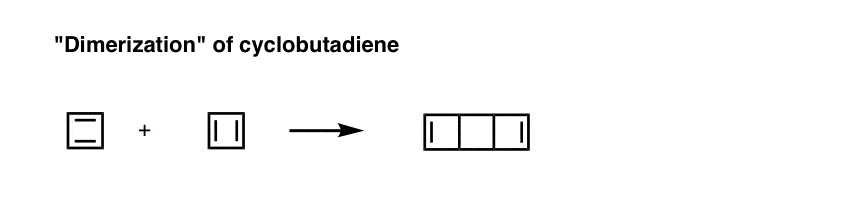
Note 2. Upon warming (–80° is considered “warm” for these purposes), cyclobutadiene reacts with itself through a Diels-Alder process to give “dimeric” species.
Note 3. If benzene is about 36 kcal/mol more stable than (theoretical) cyclohexatriene, exactly how unstable is cyclobutadiene? The negative resonance energy of cyclobutadiene is calculated to be –54.7 kcal/mol, relative to 1,3-butadiene. In addition, 30.7 kcal/mol of strain is found, giving a total destabilization of 85.4 kcal/mol. [Ref]
(Advanced) References and Further Reading
- Cyclobutadiene
Watts, J. D. Fitzpatrick, and R. Pettit
Journal of the American Chemical Society 1965, 87 (14), 3253-3254
DOI: 10.1021/ja01092a049
Interestingly, this paper precedes an article on the reactivity of cyclobutadiene-iron tricarbonyl. Cyclobutadiene is prepared by the oxidation of that organometallic complex using Ce4+, which is then trapped in situ with an alkyne. - Cyclobutadiene
Thomas Bally, Satoru Masamune
Tetrahedron 1980, 36 (3), 343-370
DOI: 1016/0040-4020(80)87003-7
This paper from 1980 reviews work done on cyclobutadiene up to that time. This is divided into 2 parts – experimental synthetic efforts and theoretical calculations.Elaborate MO treatments and theoretical calculations indicate that the most stable geometry for cyclobutadiene is rectangular. - Potential energy surfaces of cyclobutadiene: ab initio SCF and CI calculations for the low-lying singlet and triplet states
A. Jafri and M. D. Newton
Journal of the American Chemical Society 1978, 100 (16), 5012-5017
DOI: 10.1021/ja00484a016 - The potential surfaces for the lowest singlet and triplet states of cyclobutadiene
Weston Thatcher Borden, Ernest R. Davidson, and Paul Hart
Journal of the American Chemical Society 1978, 100 (2), 388-392
DOI: 10.1021/ja00470a006 - A theoretical study of the structure of cyclobutadiene
Kollmar and V. Staemmler
Journal of the American Chemical Society 1977, 99 (11), 3583-3587
DOI: 10.1021/ja00453a009 - Ground states of molecules. 36. The cyclobutadiene problem and MINDO/3 calculations of molecular vibration frequencies
Michael J. S. Dewar and Andrew Komornicki
Journal of the American Chemical Society 1977, 99 (19), 6174-6179
DOI: 10.1021/ja00461a002
Analysis of the IR spectra of the product and deuterated analogs generated from labeled precursors has confirmed the theoretical conclusion that cyclobutadiene is a rectangular molecule: - Cyclobutadiene is not square
Satoru Masamune, Fernando A. Souto-Bachiller, Takahisa Machiguchi, and John E. Bertie
Journal of the American Chemical Society 1978, 100 (15), 4889-4891
DOI: 1021/ja00483a043
Ah, for the days when papers had simple, punchy titles (Refs. 1 and 2 above). - Ab initio second-order Moller-Plesset calculation of the vibrational spectra of cyclobutadiene and its isotopic derivatives
Andes Hess Jr., P. Carsky, and L. J. Schaad
Journal of the American Chemical Society 1983, 105 (4), 695-701
DOI: 10.1021/ja00342a003 - The Dimerization of Cyclobutadiene. An ab Initio CASSCF Theoretical Study
Yi Li and and K. N. Houk
Journal of the American Chemical Society 1996, 118 (4), 880-885
DOI: 10.1021/ja921663m - Ab Initio Calculation of Resonance Energies. Benzene and Cyclobutadiene
A. Hess, Jr. and L. J. Schaad
Journal of the American Chemical Society 1983, 105 (26), 7500-7505
DOI: 10.1021/ja00364a600
A paper from the 80’s using computational methods to quantify the antiaromatic destabilization of cyclobutadiene. These authors obtain a value of -54.7 kcal/mol for the negative resonance energy of cyclobutadiene. - Experimental Determination of the Antiaromaticity of Cyclobutadiene
Ashok A. Deniz, Kevin S. Peters, Gary J. Snyder
Science 1999, 286 (5442), 1119-1122
DOI: 1126/science.286.5442.1119
This is a very rigorous paper that uses novel spectroscopic techniques to determine antiaromatic destabilization of cyclobutadiene. Relative to a hypothetical strain-less, conjugated diene reference, cyclobutadiene is destabilized by a total of 87 kcal/mol, 32 kcal/mol of which can be attributed to ring strain and 55 kcal/mol to antiaromaticity (compared with 21 kcal/mol for the aromatic stabilization of benzene). - Quantentheoretische Beiträge zum Benzolproblem
Die Elektronenkonfiguration des Benzols und verwandter Verbindungen
Erich Hückel
Zeitschrift für Physik 1931, 70, 204–286
DOI: 10.1007/BF01339530
Erich Hückel achieved recognition by elaborating, together with Peter Debye, the theory of strong electrolytes in 1923 and later by applying a simplified version of quantum theory to p-electrons in conjugated molecules, which became known as Hückel molecular orbital (HMO) theory. Although he never explicitly formulated a “4n + 2 rule”, this was obvious from his work. Hückel showed that monocyclic systems with continuous conjugation having 6, 10, 14, etc. p-electrons benefited from extra stabilization and were aromatic. But it is more accurate to refer to the “Hückel 4n + 2 p-electron rule,” rather than to “Hückel’s rule.” - A Mnemonic Device for Molecular Orbital Energies
Arthur A. Frost and Boris Musulin
J. Chem. Phys. 1953, 21, 572
DOI: 10.1063/1.1698970
The origin of the “Frost Circle” mnemonic device for determining the MO’s of electrocyclic systems.
00 General Chemistry Review
01 Bonding, Structure, and Resonance
- How Do We Know Methane (CH4) Is Tetrahedral?
- Hybrid Orbitals and Hybridization
- How To Determine Hybridization: A Shortcut
- Orbital Hybridization And Bond Strengths
- Sigma bonds come in six varieties: Pi bonds come in one
- A Key Skill: How to Calculate Formal Charge
- The Four Intermolecular Forces and How They Affect Boiling Points
- 3 Trends That Affect Boiling Points
- How To Use Electronegativity To Determine Electron Density (and why NOT to trust formal charge)
- Introduction to Resonance
- How To Use Curved Arrows To Interchange Resonance Forms
- Evaluating Resonance Forms (1) - The Rule of Least Charges
- How To Find The Best Resonance Structure By Applying Electronegativity
- Evaluating Resonance Structures With Negative Charges
- Evaluating Resonance Structures With Positive Charge
- Exploring Resonance: Pi-Donation
- Exploring Resonance: Pi-acceptors
- In Summary: Evaluating Resonance Structures
- Drawing Resonance Structures: 3 Common Mistakes To Avoid
- How to apply electronegativity and resonance to understand reactivity
- Bond Hybridization Practice
- Structure and Bonding Practice Quizzes
- Resonance Structures Practice
02 Acid Base Reactions
- Introduction to Acid-Base Reactions
- Acid Base Reactions In Organic Chemistry
- The Stronger The Acid, The Weaker The Conjugate Base
- Walkthrough of Acid-Base Reactions (3) - Acidity Trends
- Five Key Factors That Influence Acidity
- Acid-Base Reactions: Introducing Ka and pKa
- How to Use a pKa Table
- The pKa Table Is Your Friend
- A Handy Rule of Thumb for Acid-Base Reactions
- Acid Base Reactions Are Fast
- pKa Values Span 60 Orders Of Magnitude
- How Protonation and Deprotonation Affect Reactivity
- Acid Base Practice Problems
03 Alkanes and Nomenclature
- Meet the (Most Important) Functional Groups
- Condensed Formulas: Deciphering What the Brackets Mean
- Hidden Hydrogens, Hidden Lone Pairs, Hidden Counterions
- Don't Be Futyl, Learn The Butyls
- Primary, Secondary, Tertiary, Quaternary In Organic Chemistry
- Branching, and Its Affect On Melting and Boiling Points
- The Many, Many Ways of Drawing Butane
- Wedge And Dash Convention For Tetrahedral Carbon
- Common Mistakes in Organic Chemistry: Pentavalent Carbon
- Table of Functional Group Priorities for Nomenclature
- Summary Sheet - Alkane Nomenclature
- Organic Chemistry IUPAC Nomenclature Demystified With A Simple Puzzle Piece Approach
- Boiling Point Quizzes
- Organic Chemistry Nomenclature Quizzes
04 Conformations and Cycloalkanes
- Staggered vs Eclipsed Conformations of Ethane
- Conformational Isomers of Propane
- Newman Projection of Butane (and Gauche Conformation)
- Introduction to Cycloalkanes
- Geometric Isomers In Small Rings: Cis And Trans Cycloalkanes
- Calculation of Ring Strain In Cycloalkanes
- Cycloalkanes - Ring Strain In Cyclopropane And Cyclobutane
- Cyclohexane Conformations
- Cyclohexane Chair Conformation: An Aerial Tour
- How To Draw The Cyclohexane Chair Conformation
- The Cyclohexane Chair Flip
- The Cyclohexane Chair Flip - Energy Diagram
- Substituted Cyclohexanes - Axial vs Equatorial
- Ranking The Bulkiness Of Substituents On Cyclohexanes: "A-Values"
- Cyclohexane Chair Conformation Stability: Which One Is Lower Energy?
- Fused Rings - Cis-Decalin and Trans-Decalin
- Naming Bicyclic Compounds - Fused, Bridged, and Spiro
- Bredt's Rule (And Summary of Cycloalkanes)
- Newman Projection Practice
- Cycloalkanes Practice Problems
05 A Primer On Organic Reactions
- The Most Important Question To Ask When Learning a New Reaction
- Curved Arrows (for reactions)
- Nucleophiles and Electrophiles
- The Three Classes of Nucleophiles
- Nucleophilicity vs. Basicity
- What Makes A Good Nucleophile?
- What Makes A Good Leaving Group?
- 3 Factors That Stabilize Carbocations
- Equilibrium and Energy Relationships
- 7 Factors that stabilize negative charge in organic chemistry
- 7 Factors That Stabilize Positive Charge in Organic Chemistry
- What's a Transition State?
- Hammond's Postulate
- Learning Organic Chemistry Reactions: A Checklist (PDF)
- Introduction to Oxidative Cleavage Reactions
06 Free Radical Reactions
- Bond Dissociation Energies = Homolytic Cleavage
- Free Radical Reactions
- 3 Factors That Stabilize Free Radicals
- What Factors Destabilize Free Radicals?
- Bond Strengths And Radical Stability
- Free Radical Initiation: Why Is "Light" Or "Heat" Required?
- Initiation, Propagation, Termination
- Monochlorination Products Of Propane, Pentane, And Other Alkanes
- Selectivity In Free Radical Reactions
- Selectivity in Free Radical Reactions: Bromination vs. Chlorination
- Halogenation At Tiffany's
- Allylic Bromination
- Bonus Topic: Allylic Rearrangements
- In Summary: Free Radicals
- Synthesis (2) - Reactions of Alkanes
- Free Radicals Practice Quizzes
07 Stereochemistry and Chirality
- Types of Isomers: Constitutional Isomers, Stereoisomers, Enantiomers, and Diastereomers
- How To Draw The Enantiomer Of A Chiral Molecule
- How To Draw A Bond Rotation
- Introduction to Assigning (R) and (S): The Cahn-Ingold-Prelog Rules
- Assigning Cahn-Ingold-Prelog (CIP) Priorities (2) - The Method of Dots
- Enantiomers vs Diastereomers vs The Same? Two Methods For Solving Problems
- Assigning R/S To Newman Projections (And Converting Newman To Line Diagrams)
- How To Determine R and S Configurations On A Fischer Projection
- The Meso Trap
- Optical Rotation, Optical Activity, and Specific Rotation
- Optical Purity and Enantiomeric Excess
- What's a Racemic Mixture?
- Chiral Allenes And Chiral Axes
- Stereochemistry Practice Problems and Quizzes
08 Substitution Reactions
- Nucleophilic Substitution Reactions - Introduction
- Two Types of Nucleophilic Substitution Reactions
- The SN2 Mechanism
- Why the SN2 Reaction Is Powerful
- The SN1 Mechanism
- The Conjugate Acid Is A Better Leaving Group
- Comparing the SN1 and SN2 Reactions
- Polar Protic? Polar Aprotic? Nonpolar? All About Solvents
- Steric Hindrance is Like a Fat Goalie
- Common Blind Spot: Intramolecular Reactions
- Substitution Practice - SN1
- Substitution Practice - SN2
09 Elimination Reactions
- Elimination Reactions (1): Introduction And The Key Pattern
- Elimination Reactions (2): The Zaitsev Rule
- Elimination Reactions Are Favored By Heat
- Two Elimination Reaction Patterns
- The E1 Reaction
- The E2 Mechanism
- E1 vs E2: Comparing the E1 and E2 Reactions
- Antiperiplanar Relationships: The E2 Reaction and Cyclohexane Rings
- Bulky Bases in Elimination Reactions
- Comparing the E1 vs SN1 Reactions
- Elimination (E1) Reactions With Rearrangements
- E1cB - Elimination (Unimolecular) Conjugate Base
- Elimination (E1) Practice Problems And Solutions
- Elimination (E2) Practice Problems and Solutions
10 Rearrangements
11 SN1/SN2/E1/E2 Decision
- Identifying Where Substitution and Elimination Reactions Happen
- Deciding SN1/SN2/E1/E2 (1) - The Substrate
- Deciding SN1/SN2/E1/E2 (2) - The Nucleophile/Base
- SN1 vs E1 and SN2 vs E2 : The Temperature
- Deciding SN1/SN2/E1/E2 - The Solvent
- Wrapup: The Key Factors For Determining SN1/SN2/E1/E2
- Alkyl Halide Reaction Map And Summary
- SN1 SN2 E1 E2 Practice Problems
12 Alkene Reactions
- E and Z Notation For Alkenes (+ Cis/Trans)
- Alkene Stability
- Alkene Addition Reactions: "Regioselectivity" and "Stereoselectivity" (Syn/Anti)
- Stereoselective and Stereospecific Reactions
- Hydrohalogenation of Alkenes and Markovnikov's Rule
- Hydration of Alkenes With Aqueous Acid
- Rearrangements in Alkene Addition Reactions
- Halogenation of Alkenes and Halohydrin Formation
- Oxymercuration Demercuration of Alkenes
- Hydroboration Oxidation of Alkenes
- m-CPBA (meta-chloroperoxybenzoic acid)
- OsO4 (Osmium Tetroxide) for Dihydroxylation of Alkenes
- Palladium on Carbon (Pd/C) for Catalytic Hydrogenation of Alkenes
- Cyclopropanation of Alkenes
- A Fourth Alkene Addition Pattern - Free Radical Addition
- Alkene Reactions: Ozonolysis
- Summary: Three Key Families Of Alkene Reaction Mechanisms
- Synthesis (4) - Alkene Reaction Map, Including Alkyl Halide Reactions
- Alkene Reactions Practice Problems
13 Alkyne Reactions
- Acetylides from Alkynes, And Substitution Reactions of Acetylides
- Partial Reduction of Alkynes With Lindlar's Catalyst
- Partial Reduction of Alkynes With Na/NH3 To Obtain Trans Alkenes
- Alkyne Hydroboration With "R2BH"
- Hydration and Oxymercuration of Alkynes
- Hydrohalogenation of Alkynes
- Alkyne Halogenation: Bromination, Chlorination, and Iodination of Alkynes
- Alkyne Reactions - The "Concerted" Pathway
- Alkenes To Alkynes Via Halogenation And Elimination Reactions
- Alkynes Are A Blank Canvas
- Synthesis (5) - Reactions of Alkynes
- Alkyne Reactions Practice Problems With Answers
14 Alcohols, Epoxides and Ethers
- Alcohols - Nomenclature and Properties
- Alcohols Can Act As Acids Or Bases (And Why It Matters)
- Alcohols - Acidity and Basicity
- The Williamson Ether Synthesis
- Ethers From Alkenes, Tertiary Alkyl Halides and Alkoxymercuration
- Alcohols To Ethers via Acid Catalysis
- Cleavage Of Ethers With Acid
- Epoxides - The Outlier Of The Ether Family
- Opening of Epoxides With Acid
- Epoxide Ring Opening With Base
- Making Alkyl Halides From Alcohols
- Tosylates And Mesylates
- PBr3 and SOCl2
- Elimination Reactions of Alcohols
- Elimination of Alcohols To Alkenes With POCl3
- Alcohol Oxidation: "Strong" and "Weak" Oxidants
- Demystifying The Mechanisms of Alcohol Oxidations
- Protecting Groups For Alcohols
- Thiols And Thioethers
- Calculating the oxidation state of a carbon
- Oxidation and Reduction in Organic Chemistry
- Oxidation Ladders
- SOCl2 Mechanism For Alcohols To Alkyl Halides: SN2 versus SNi
- Alcohol Reactions Roadmap (PDF)
- Alcohol Reaction Practice Problems
- Epoxide Reaction Quizzes
- Oxidation and Reduction Practice Quizzes
15 Organometallics
- What's An Organometallic?
- Formation of Grignard and Organolithium Reagents
- Organometallics Are Strong Bases
- Reactions of Grignard Reagents
- Protecting Groups In Grignard Reactions
- Synthesis Problems Involving Grignard Reagents
- Grignard Reactions And Synthesis (2)
- Organocuprates (Gilman Reagents): How They're Made
- Gilman Reagents (Organocuprates): What They're Used For
- The Heck, Suzuki, and Olefin Metathesis Reactions (And Why They Don't Belong In Most Introductory Organic Chemistry Courses)
- Reaction Map: Reactions of Organometallics
- Grignard Practice Problems
16 Spectroscopy
- Degrees of Unsaturation (or IHD, Index of Hydrogen Deficiency)
- Conjugation And Color (+ How Bleach Works)
- Introduction To UV-Vis Spectroscopy
- UV-Vis Spectroscopy: Absorbance of Carbonyls
- UV-Vis Spectroscopy: Practice Questions
- Bond Vibrations, Infrared Spectroscopy, and the "Ball and Spring" Model
- Infrared Spectroscopy: A Quick Primer On Interpreting Spectra
- IR Spectroscopy: 4 Practice Problems
- 1H NMR: How Many Signals?
- Homotopic, Enantiotopic, Diastereotopic
- Diastereotopic Protons in 1H NMR Spectroscopy: Examples
- 13-C NMR - How Many Signals
- Liquid Gold: Pheromones In Doe Urine
- Natural Product Isolation (1) - Extraction
- Natural Product Isolation (2) - Purification Techniques, An Overview
- Structure Determination Case Study: Deer Tarsal Gland Pheromone
17 Dienes and MO Theory
- What To Expect In Organic Chemistry 2
- Are these molecules conjugated?
- Conjugation And Resonance In Organic Chemistry
- Bonding And Antibonding Pi Orbitals
- Molecular Orbitals of The Allyl Cation, Allyl Radical, and Allyl Anion
- Pi Molecular Orbitals of Butadiene
- Reactions of Dienes: 1,2 and 1,4 Addition
- Thermodynamic and Kinetic Products
- More On 1,2 and 1,4 Additions To Dienes
- s-cis and s-trans
- The Diels-Alder Reaction
- Cyclic Dienes and Dienophiles in the Diels-Alder Reaction
- Stereochemistry of the Diels-Alder Reaction
- Exo vs Endo Products In The Diels Alder: How To Tell Them Apart
- HOMO and LUMO In the Diels Alder Reaction
- Why Are Endo vs Exo Products Favored in the Diels-Alder Reaction?
- Diels-Alder Reaction: Kinetic and Thermodynamic Control
- The Retro Diels-Alder Reaction
- The Intramolecular Diels Alder Reaction
- Regiochemistry In The Diels-Alder Reaction
- The Cope and Claisen Rearrangements
- Electrocyclic Reactions
- Electrocyclic Ring Opening And Closure (2) - Six (or Eight) Pi Electrons
- Diels Alder Practice Problems
- Molecular Orbital Theory Practice
18 Aromaticity
- Introduction To Aromaticity
- Rules For Aromaticity
- Huckel's Rule: What Does 4n+2 Mean?
- Aromatic, Non-Aromatic, or Antiaromatic? Some Practice Problems
- Antiaromatic Compounds and Antiaromaticity
- The Pi Molecular Orbitals of Benzene
- The Pi Molecular Orbitals of Cyclobutadiene
- Frost Circles
- Aromaticity Practice Quizzes
19 Reactions of Aromatic Molecules
- Electrophilic Aromatic Substitution: Introduction
- Activating and Deactivating Groups In Electrophilic Aromatic Substitution
- Electrophilic Aromatic Substitution - The Mechanism
- Ortho-, Para- and Meta- Directors in Electrophilic Aromatic Substitution
- Understanding Ortho, Para, and Meta Directors
- Why are halogens ortho- para- directors?
- Disubstituted Benzenes: The Strongest Electron-Donor "Wins"
- Electrophilic Aromatic Substitutions (1) - Halogenation of Benzene
- Electrophilic Aromatic Substitutions (2) - Nitration and Sulfonation
- EAS Reactions (3) - Friedel-Crafts Acylation and Friedel-Crafts Alkylation
- Intramolecular Friedel-Crafts Reactions
- Nucleophilic Aromatic Substitution (NAS)
- Nucleophilic Aromatic Substitution (2) - The Benzyne Mechanism
- Reactions on the "Benzylic" Carbon: Bromination And Oxidation
- The Wolff-Kishner, Clemmensen, And Other Carbonyl Reductions
- More Reactions on the Aromatic Sidechain: Reduction of Nitro Groups and the Baeyer Villiger
- Aromatic Synthesis (1) - "Order Of Operations"
- Synthesis of Benzene Derivatives (2) - Polarity Reversal
- Aromatic Synthesis (3) - Sulfonyl Blocking Groups
- Birch Reduction
- Synthesis (7): Reaction Map of Benzene and Related Aromatic Compounds
- Aromatic Reactions and Synthesis Practice
- Electrophilic Aromatic Substitution Practice Problems
20 Aldehydes and Ketones
- What's The Alpha Carbon In Carbonyl Compounds?
- Nucleophilic Addition To Carbonyls
- Aldehydes and Ketones: 14 Reactions With The Same Mechanism
- Sodium Borohydride (NaBH4) Reduction of Aldehydes and Ketones
- Grignard Reagents For Addition To Aldehydes and Ketones
- Wittig Reaction
- Hydrates, Hemiacetals, and Acetals
- Imines - Properties, Formation, Reactions, and Mechanisms
- All About Enamines
- Breaking Down Carbonyl Reaction Mechanisms: Reactions of Anionic Nucleophiles (Part 2)
- Aldehydes Ketones Reaction Practice
21 Carboxylic Acid Derivatives
- Nucleophilic Acyl Substitution (With Negatively Charged Nucleophiles)
- Addition-Elimination Mechanisms With Neutral Nucleophiles (Including Acid Catalysis)
- Basic Hydrolysis of Esters - Saponification
- Transesterification
- Proton Transfer
- Fischer Esterification - Carboxylic Acid to Ester Under Acidic Conditions
- Lithium Aluminum Hydride (LiAlH4) For Reduction of Carboxylic Acid Derivatives
- LiAlH[Ot-Bu]3 For The Reduction of Acid Halides To Aldehydes
- Di-isobutyl Aluminum Hydride (DIBAL) For The Partial Reduction of Esters and Nitriles
- Amide Hydrolysis
- Thionyl Chloride (SOCl2)
- Diazomethane (CH2N2)
- Carbonyl Chemistry: Learn Six Mechanisms For the Price Of One
- Making Music With Mechanisms (PADPED)
- Carboxylic Acid Derivatives Practice Questions
22 Enols and Enolates
- Keto-Enol Tautomerism
- Enolates - Formation, Stability, and Simple Reactions
- Kinetic Versus Thermodynamic Enolates
- Aldol Addition and Condensation Reactions
- Reactions of Enols - Acid-Catalyzed Aldol, Halogenation, and Mannich Reactions
- Claisen Condensation and Dieckmann Condensation
- Decarboxylation
- The Malonic Ester and Acetoacetic Ester Synthesis
- The Michael Addition Reaction and Conjugate Addition
- The Robinson Annulation
- Haloform Reaction
- The Hell–Volhard–Zelinsky Reaction
- Enols and Enolates Practice Quizzes
23 Amines
- The Amide Functional Group: Properties, Synthesis, and Nomenclature
- Basicity of Amines And pKaH
- 5 Key Basicity Trends of Amines
- The Mesomeric Effect And Aromatic Amines
- Nucleophilicity of Amines
- Alkylation of Amines (Sucks!)
- Reductive Amination
- The Gabriel Synthesis
- Some Reactions of Azides
- The Hofmann Elimination
- The Hofmann and Curtius Rearrangements
- The Cope Elimination
- Protecting Groups for Amines - Carbamates
- The Strecker Synthesis of Amino Acids
- Introduction to Peptide Synthesis
- Reactions of Diazonium Salts: Sandmeyer and Related Reactions
- Amine Practice Questions
24 Carbohydrates
- D and L Notation For Sugars
- Pyranoses and Furanoses: Ring-Chain Tautomerism In Sugars
- What is Mutarotation?
- Reducing Sugars
- The Big Damn Post Of Carbohydrate-Related Chemistry Definitions
- The Haworth Projection
- Converting a Fischer Projection To A Haworth (And Vice Versa)
- Reactions of Sugars: Glycosylation and Protection
- The Ruff Degradation and Kiliani-Fischer Synthesis
- Isoelectric Points of Amino Acids (and How To Calculate Them)
- Carbohydrates Practice
- Amino Acid Quizzes
25 Fun and Miscellaneous
- A Gallery of Some Interesting Molecules From Nature
- Screw Organic Chemistry, I'm Just Going To Write About Cats
- On Cats, Part 1: Conformations and Configurations
- On Cats, Part 2: Cat Line Diagrams
- On Cats, Part 4: Enantiocats
- On Cats, Part 6: Stereocenters
- Organic Chemistry Is Shit
- The Organic Chemistry Behind "The Pill"
- Maybe they should call them, "Formal Wins" ?
- Why Do Organic Chemists Use Kilocalories?
- The Principle of Least Effort
- Organic Chemistry GIFS - Resonance Forms
- Reproducibility In Organic Chemistry
- What Holds The Nucleus Together?
- How Reactions Are Like Music
- Organic Chemistry and the New MCAT
26 Organic Chemistry Tips and Tricks
- Common Mistakes: Formal Charges Can Mislead
- Partial Charges Give Clues About Electron Flow
- Draw The Ugly Version First
- Organic Chemistry Study Tips: Learn the Trends
- The 8 Types of Arrows In Organic Chemistry, Explained
- Top 10 Skills To Master Before An Organic Chemistry 2 Final
- Common Mistakes with Carbonyls: Carboxylic Acids... Are Acids!
- Planning Organic Synthesis With "Reaction Maps"
- Alkene Addition Pattern #1: The "Carbocation Pathway"
- Alkene Addition Pattern #2: The "Three-Membered Ring" Pathway
- Alkene Addition Pattern #3: The "Concerted" Pathway
- Number Your Carbons!
- The 4 Major Classes of Reactions in Org 1
- How (and why) electrons flow
- Grossman's Rule
- Three Exam Tips
- A 3-Step Method For Thinking Through Synthesis Problems
- Putting It Together
- Putting Diels-Alder Products in Perspective
- The Ups and Downs of Cyclohexanes
- The Most Annoying Exceptions in Org 1 (Part 1)
- The Most Annoying Exceptions in Org 1 (Part 2)
- The Marriage May Be Bad, But the Divorce Still Costs Money
- 9 Nomenclature Conventions To Know
- Nucleophile attacks Electrophile
27 Case Studies of Successful O-Chem Students
- Success Stories: How Corina Got The The "Hard" Professor - And Got An A+ Anyway
- How Helena Aced Organic Chemistry
- From a "Drop" To B+ in Org 2 – How A Hard Working Student Turned It Around
- How Serge Aced Organic Chemistry
- Success Stories: How Zach Aced Organic Chemistry 1
- Success Stories: How Kari Went From C– to B+
- How Esther Bounced Back From a "C" To Get A's In Organic Chemistry 1 And 2
- How Tyrell Got The Highest Grade In Her Organic Chemistry Course
- This Is Why Students Use Flashcards
- Success Stories: How Stu Aced Organic Chemistry
- How John Pulled Up His Organic Chemistry Exam Grades
- Success Stories: How Nathan Aced Organic Chemistry (Without It Taking Over His Life)
- How Chris Aced Org 1 and Org 2
- Interview: How Jay Got an A+ In Organic Chemistry
- How to Do Well in Organic Chemistry: One Student's Advice
- "America's Top TA" Shares His Secrets For Teaching O-Chem
- "Organic Chemistry Is Like..." - A Few Metaphors
- How To Do Well In Organic Chemistry: Advice From A Tutor
- Guest post: "I went from being afraid of tests to actually looking forward to them".
sir please draw the digram of carbon 7 pi molecular orbital picture
Cyclic or acyclic?
Hello!
I’m confused as to where to locate the Pi bonds when looking at the MO orbital. Since the two valence orbitals in cyclobutadiene are in non bonding orbitals and are singly occupied, why then do we see two pi bonds? I understand how Pi 1 represents a double bond but I don’t see where the second bond is. Thank you!
Hi Danielle – You can think of the situation where both carbons have free radicals as a “resonance form” of a pi bond.
(my answer here could, and should, go a lot deeper, but I’m just going to keep it simple in this case).